Posts Tagged ‘Organic chemistry’
Monday, February 18th, 2019
Students learning organic chemistry are often asked in examinations and tutorials to devise the mechanisms (as represented by curly arrows) for the core corpus of important reactions, with the purpose of learning skills that allow them to go on to improvise mechanisms for new reactions. A common question asked by students is how should such mechanisms be presented in an exam in order to gain full credit? Alternatively, is there a single correct mechanism for any given reaction? To which the lecturer or tutor will often respond that any reasonable mechanism will receive such credit. The implication is that a mechanism is “reasonable” if it “follows the rules”. The rules are rarely declared fully, but seem to be part of the absorbed but often mysterious skill acquired in learning the subject. These rules also include those governing how the curly arrows should be drawn.† Here I explore this topic using the Graham reaction.[1]‡
(more…)
References
- W.H. Graham, "The Halogenation of Amidines. I. Synthesis of 3-Halo- and Other Negatively Substituted Diazirines<sup>1</sup>", Journal of the American Chemical Society, vol. 87, pp. 4396-4397, 1965. https://doi.org/10.1021/ja00947a040
Tags:/RT, activation energy, activation free energy, animation, arrow pushing, arrow-head, cellular telephone, Chemical kinetics, chemical reaction, Chemistry, computed energy, Ed Smith, energy, energy maximum, energy minima, energy plot, energy profile, energy surface, free energy, lecturer, mechanism, Natural sciences, Organic chemistry, overall reaction energy, Physical sciences, Reaction rate constant, Resonance, Transition state, Transition state theory, tutor, Tutorial
Posted in Curly arrows, Interesting chemistry | No Comments »
Sunday, January 13th, 2019
Linear free energy relationships (LFER) are associated with the dawn of physical organic chemistry in the late 1930s and its objectives in understanding chemical reactivity as measured by reaction rates and equilibria.
(more…)
Tags:Benzoic acid, Chemical kinetics, chemical reaction, chemical reactivity, chemist, Chemistry, Electrophilic aromatic substitution, energy point, Equations, Equilibrium chemistry, Equilibrium constant, free energy overall route, Hammett equation, Linear free energy relationships, Natural sciences, Organic chemistry, Physical organic chemistry, Physical sciences, Reactivity
Posted in Chemical IT, Interesting chemistry, reaction mechanism | No Comments »
Friday, December 21st, 2018
Five years back, I speculated about the mechanism of the epoxidation of ethene by a peracid, concluding that kinetic isotope effects provided interesting evidence that this mechanism is highly asynchronous and involves a so-called “hidden intermediate”. Here I revisit this reaction in which a small change is applied to the atoms involved.
(more…)
Tags:Chemical kinetics, chemical reaction, Chemistry, Deuterium, Isotope effect, Kinetic isotope effect, Natural sciences, Organic chemistry, overall activation energy, pericyclic reaction, Physical organic chemistry, Physical sciences, potential energy surface, Rearrangement reactions
Posted in Interesting chemistry | 5 Comments »
Sunday, August 26th, 2018
In the previous post, I investigated the mechanism of cyclopropanation of an enal using a benzylic chloride using a quantum chemistry based procedure. Here I take a look at the NMR spectra of the resulting cyclopropane products, with an evaluation of the original stereochemical assignments.[1]
(more…)
References
- M. Meazza, A. Kowalczuk, S. Watkins, S. Holland, T.A. Logothetis, and R. Rios, "Organocatalytic Cyclopropanation of (<i>E</i>)-Dec-2-enal: Synthesis, Spectral Analysis and Mechanistic Understanding", Journal of Chemical Education, vol. 95, pp. 1832-1839, 2018. https://doi.org/10.1021/acs.jchemed.7b00566
Tags:Benzyl group, Chemistry, Cyclopropanation, cyclopropane products, Cyclopropanes, Nuclear magnetic resonance, Organic chemistry, Organic reactions, Protecting groups
Posted in Interesting chemistry | No Comments »
Saturday, August 25th, 2018
Symbiosis between computation and experiment is increasingly evident in pedagogic journals such as J. Chemical Education. Thus an example of original laboratory experiments[1],[2] that later became twinned with a computational counterpart.[3] So when I spotted this recent lab experiment[4] I felt another twinning approaching.
(more…)
References
- A. Burke, P. Dillon, K. Martin, and T.W. Hanks, "Catalytic Asymmetric Epoxidation Using a Fructose-Derived Catalyst", Journal of Chemical Education, vol. 77, pp. 271, 2000. https://doi.org/10.1021/ed077p271
- J. Hanson, "Synthesis and Use of Jacobsen's Catalyst: Enantioselective Epoxidation in the Introductory Organic Laboratory", Journal of Chemical Education, vol. 78, pp. 1266, 2001. https://doi.org/10.1021/ed078p1266
- K.K.(. Hii, H.S. Rzepa, and E.H. Smith, "Asymmetric Epoxidation: A Twinned Laboratory and Molecular Modeling Experiment for Upper-Level Organic Chemistry Students", Journal of Chemical Education, vol. 92, pp. 1385-1389, 2015. https://doi.org/10.1021/ed500398e
- M. Meazza, A. Kowalczuk, S. Watkins, S. Holland, T.A. Logothetis, and R. Rios, "Organocatalytic Cyclopropanation of (<i>E</i>)-Dec-2-enal: Synthesis, Spectral Analysis and Mechanistic Understanding", Journal of Chemical Education, vol. 95, pp. 1832-1839, 2018. https://doi.org/10.1021/acs.jchemed.7b00566
Tags:Ammonium, Benzyl group, Cations, chemical diagrams, Chemistry, condensation, final product, Functional groups, Iminium, Methyl group, Name reactions, Organic chemistry, possible diastereomeric products, relative energy, Vector Graphics, web browsers
Posted in Interesting chemistry | 9 Comments »
Wednesday, August 8th, 2018
White City is a small area in west london created as an exhibition site in 1908, morphing over the years into an Olympic games venue, a greyhound track, the home nearby of the BBC (British Broadcasting Corporation) and most recently the new western campus for Imperial College London.♣ The first Imperial department to move into the MSRH (Molecular Sciences Research Hub) building is chemistry. As a personal celebration of this occasion, I here dedicate three transition states located during my first week of occupancy there, naming them the White City trio following earlier inspiration by a string trio and their own instruments.
(more…)
Tags:acetic acid, Acid, Amide, Amine, carboxylic acid, Chemistry, Company: BBC, Company: British Broadcasting Corporation, energy, Ester, exhibition site, free energy barrier, Functional groups, Hydrogen bond, Imperial College, Imperial College London, Ionic product, Newspaper & Magazine Printing Services, Non-ionic product, Olympic games, Organic chemistry, White City Trio
Posted in Interesting chemistry | 6 Comments »
Wednesday, March 7th, 2018
C&EN has again run a vote for the 2017 Molecules of the year. Here I take a look not just at these molecules, but at how FAIR (Findable, Accessible, Interoperable and Reusable) the data associated with these molecules actually is.
(more…)
Tags:Carotenoids, Chemistry, Epoxides, Macrocycles, Organic chemistry, Organofluorides, PDF, Peptides, search engine, search program, search.datacite.org search engine, Technology/Internet
Posted in Chemical IT, crystal_structure_mining, Interesting chemistry | No Comments »
Tuesday, December 26th, 2017
Recollect the suggestion that diazomethane has hypervalent character[1]. When I looked into this, I came to the conclusion that it probably was mildly hypervalent, but on carbon and not nitrogen. Here I try some variations with substituents to see what light if any this casts.

(more…)
References
- M.C. Durrant, "A quantitative definition of hypervalency", Chemical Science, vol. 6, pp. 6614-6623, 2015. https://doi.org/10.1039/c5sc02076j
Tags:chemical bonding, Chemistry, diazo, Diazo compounds, Diazomethane, diazomethane-like systems, Functional groups, Hypervalent molecule, Molecular geometry, Organic chemistry, Recollects
Posted in Hypervalency | 8 Comments »
Sunday, October 1st, 2017
I noted in my WATOC conference report a presentation describing the use of calculated reaction barriers (and derived rate constants) as mechanistic reality checks. Computations, it was claimed, have now reached a level of accuracy whereby a barrier calculated as being 6 kcal/mol too high can start ringing mechanistic alarm bells. So when I came across this article[1] in which calculated barriers for a dyotropic ring expansion observed under mild conditions in dichloromethane as solvent were used to make mechanistic inferences, I decided to explore the mechanism a bit further.
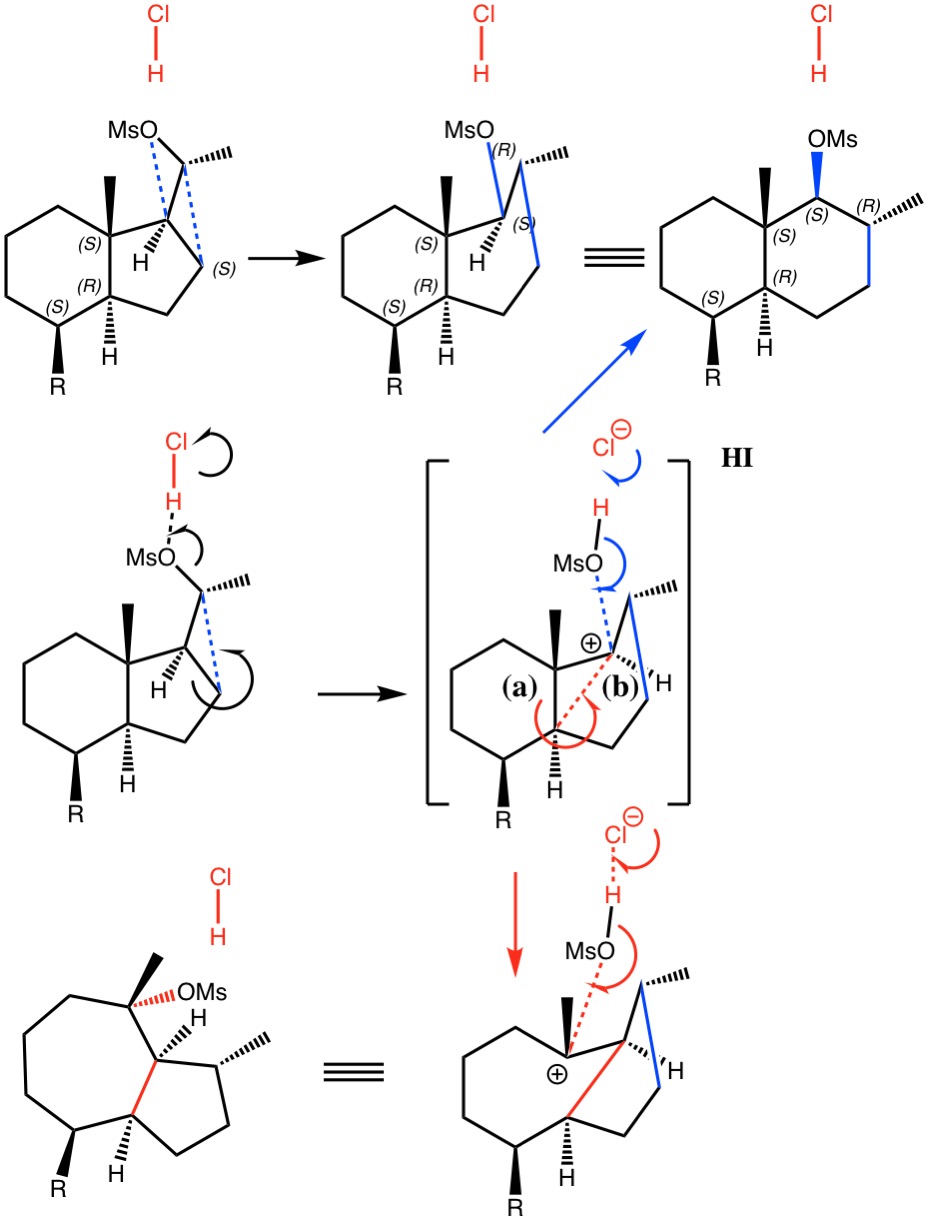
(more…)
References
- H. Santalla, O.N. Faza, G. Gómez, Y. Fall, and C. Silva López, "From Hydrindane to Decalin: A Mild Transformation through a Dyotropic Ring Expansion", Organic Letters, vol. 19, pp. 3648-3651, 2017. https://doi.org/10.1021/acs.orglett.7b01621
Tags:animation, bicyclic ring product, energy derivative gradient norm, energy profile, final non-ionic product, Organic chemistry, possible products, potential energy surface, realistic model for the reaction
Posted in pericyclic, reaction mechanism | 3 Comments »
Thursday, April 13th, 2017
Layer stacking in structures such as graphite is well-studied. The separation between the π-π planes is ~3.35Å, which is close to twice the estimated van der Waals (vdW) radius of carbon (1.7Å). But how much closer could such layers get, given that many other types of relatively weak interaction such as hydrogen bonding can contract the vdW distance sum by up to ~0.8Å or even more? This question was prompted by the separation calculated for the ion-pair cyclopropenium cyclopentadienide (~2.6-2.8Å).
(more…)
Tags:Carbon, chemical bonding, Chemistry, Cyclopentadienyl anion, Graphite, Hydrogen bond, Intermolecular forces, Nature, Organic chemistry, search query, Stacking, Supramolecular chemistry, VDW
Posted in crystal_structure_mining | 1 Comment »