Posts Tagged ‘activation energy’
Monday, February 18th, 2019
Students learning organic chemistry are often asked in examinations and tutorials to devise the mechanisms (as represented by curly arrows) for the core corpus of important reactions, with the purpose of learning skills that allow them to go on to improvise mechanisms for new reactions. A common question asked by students is how should such mechanisms be presented in an exam in order to gain full credit? Alternatively, is there a single correct mechanism for any given reaction? To which the lecturer or tutor will often respond that any reasonable mechanism will receive such credit. The implication is that a mechanism is “reasonable” if it “follows the rules”. The rules are rarely declared fully, but seem to be part of the absorbed but often mysterious skill acquired in learning the subject. These rules also include those governing how the curly arrows should be drawn.† Here I explore this topic using the Graham reaction.[1]‡
(more…)
References
- W.H. Graham, "The Halogenation of Amidines. I. Synthesis of 3-Halo- and Other Negatively Substituted Diazirines<sup>1</sup>", Journal of the American Chemical Society, vol. 87, pp. 4396-4397, 1965. https://doi.org/10.1021/ja00947a040
Tags:/RT, activation energy, activation free energy, animation, arrow pushing, arrow-head, cellular telephone, Chemical kinetics, chemical reaction, Chemistry, computed energy, Ed Smith, energy, energy maximum, energy minima, energy plot, energy profile, energy surface, free energy, lecturer, mechanism, Natural sciences, Organic chemistry, overall reaction energy, Physical sciences, Reaction rate constant, Resonance, Transition state, Transition state theory, tutor, Tutorial
Posted in Curly arrows, Interesting chemistry | No Comments »
Monday, November 23rd, 2015
This reaction emerged a few years ago (thanks Alan!) as a tutorial problem in organic chemistry, in which students had to devise a mechanism for the reaction and use this to predict the stereochemical outcome at the two chiral centres indicated with *. It originates in a brief report from R. B. Woodward’s group in 1973 describing a prostaglandin synthesis,[1] the stereochemical outcome being crucial. Here I take a look at this mechanism using computation.
(more…)
References
- R.B. Woodward, J. Gosteli, I. Ernest, R.J. Friary, G. Nestler, H. Raman, R. Sitrin, C. Suter, and J.K. Whitesell, "Novel synthesis of prostaglandin F2.alpha.", Journal of the American Chemical Society, vol. 95, pp. 6853-6855, 1973. https://doi.org/10.1021/ja00801a066
Tags:activation energy, animation, energy, learning tool
Posted in Interesting chemistry, reaction mechanism | 4 Comments »
Monday, August 18th, 2014
This post, the fifth in the series, comes full circle. I started off by speculating how to invert the stereochemical outcome of an electrocyclic reaction by inverting a bond polarity. This led to finding transition states for BOTH outcomes with suitable substitution, and then seeking other examples. Migration in homotropylium cation was one such, with the “allowed/retention” transition state proving a (little) lower in activation energy than the “forbidden/inversion” path. Here, I show that with two electrons less, the stereochemical route indeed inverts. First, a [1,4] alkyl shift with inversion at the migrating carbon (ωB97XD/6-311G(d,p)/SCRF=chloroform); as a four-electron process, this is the “allowed” route.[1]
First, a [1,4] alkyl shift with inversion at the migrating carbon (ωB97XD/6-311G(d,p)/SCRF=chloroform); as a four-electron process, this is the “allowed” route.[1] 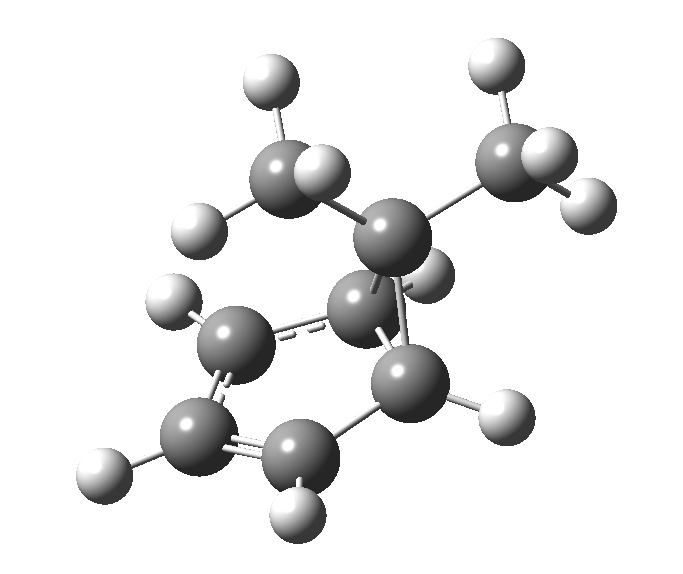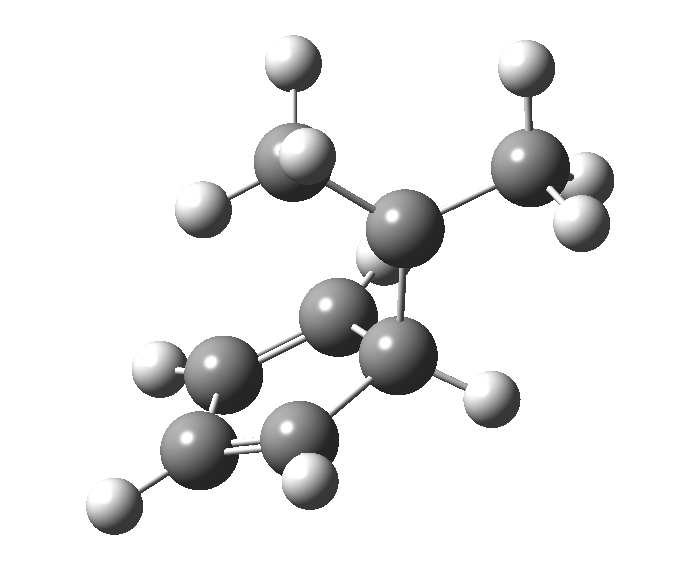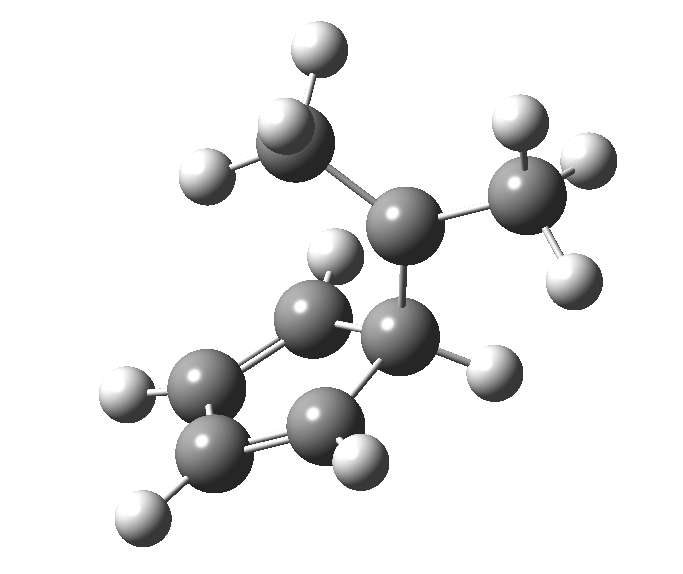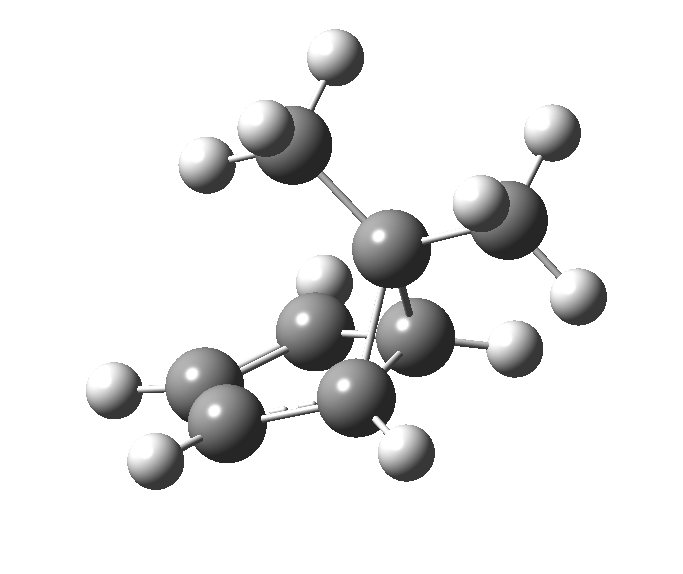 The “forbidden” route corresponds to retention of configuration at the migrating carbon.[2]
The “forbidden” route corresponds to retention of configuration at the migrating carbon.[2] 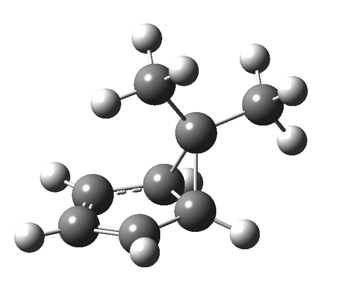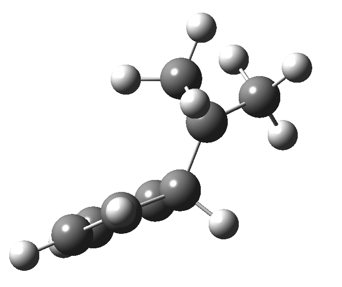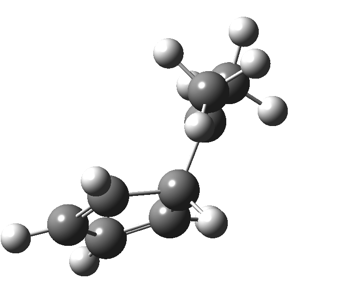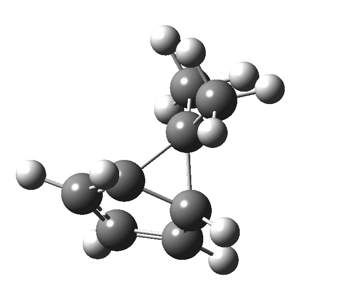 The barriers for each process can be seen below from the IRCs. That for inversion is ~4.5 kcal/mol lower than retention. This nicely transposes the values for the six-electron homologue shown in the previous post.
The barriers for each process can be seen below from the IRCs. That for inversion is ~4.5 kcal/mol lower than retention. This nicely transposes the values for the six-electron homologue shown in the previous post. 
 There is one more nugget of insight that can be extracted. The start/end-point for the six-electron process (homotropylium cation) was, as the name implies, homoaromatic. Now, with a four-electron system we also have an inverse. Nominally, we should now end with homo-antiaromaticity (but see [3]). But antiaromaticity is avoided whenever possible, and so the homoaromatic bond observed in homotropylium is not formed. It resolutely remains a σ-bond (1.48Å) thus sequestering two electrons, and the remaining two electrons simply form a delocalised allyl cation. With the six-electron homotropylium, reactant/product were stabilised by that additional (homo)aromaticity, thus inducing a relatively high barrier. With the four-electron system here, no such reactant/product stabilisation occurs, and hence the reaction barriers are now significantly lower. A rather neat pedagogic example.
There is one more nugget of insight that can be extracted. The start/end-point for the six-electron process (homotropylium cation) was, as the name implies, homoaromatic. Now, with a four-electron system we also have an inverse. Nominally, we should now end with homo-antiaromaticity (but see [3]). But antiaromaticity is avoided whenever possible, and so the homoaromatic bond observed in homotropylium is not formed. It resolutely remains a σ-bond (1.48Å) thus sequestering two electrons, and the remaining two electrons simply form a delocalised allyl cation. With the six-electron homotropylium, reactant/product were stabilised by that additional (homo)aromaticity, thus inducing a relatively high barrier. With the four-electron system here, no such reactant/product stabilisation occurs, and hence the reaction barriers are now significantly lower. A rather neat pedagogic example.
References
- H.S. Rzepa, "Gaussian Job Archive for C8H11(1+)", 2014. https://doi.org/10.6084/m9.figshare.1142175
- H.S. Rzepa, "Gaussian Job Archive for C8H11(1+)", 2014. https://doi.org/10.6084/m9.figshare.1142174
- C.S.M. Allan, and H.S. Rzepa, "Chiral Aromaticities. A Topological Exploration of Möbius Homoaromaticity", Journal of Chemical Theory and Computation, vol. 4, pp. 1841-1848, 2008. https://doi.org/10.1021/ct8001915
Tags:activation energy, reactant/product
Posted in pericyclic, reaction mechanism | 2 Comments »
Wednesday, March 12th, 2014
My previous post related to the aromatic electrophilic substitution of benzene using as electrophile phenyl diazonium chloride. Another prototypical reaction, and again one where benzene is too inactive for the reaction to occur easily, is the catalyst-free bromination of benzene to give bromobenzene and HBr.
(more…)
Tags:activation energy, animation, aromatic, Boris Galabov, co-author, electrophilic, lowest energy solution, o/p director of aromatic electrophilic substitution, Paul Schleyer, pence, substitution
Posted in Interesting chemistry, reaction mechanism | 8 Comments »
Sunday, October 16th, 2011
I asked a while back whether blogs could be considered a serious form of scholarly scientific communication (and so has Peter Murray-Rust more recently). A case for doing so might be my post of about a year ago, addressing why borane reduces a carboxylic acid, but not its ester, where I suggested a possible mechanism. Well, colleagues have raised some interesting questions, both on the blog itself and more silently by email to me. As a result, I have tried to address some of these questions, and accordingly my original scheme needs some revision! This sort of iterative process of getting to the truth with the help of the community (a kind of crowd-sourced chemistry) is where I feel blogs do have a genuine role to play.

The reduction of a carboxylic acid by borane
(more…)
Tags:activation energy, animation, carboxylic acid, carboxylic ester, diborane, free energy, pericyclic, Peter Murray-Rust, Reaction Mechanism, reduction, Tutorial material
Posted in Interesting chemistry | 8 Comments »
