The diazo-coupling reaction dates back to the 1850s (and a close association with Imperial College via the first professor of chemistry there, August von Hofmann) and its mechanism was much studied in the heyday of physical organic chemistry.[1] Nick Greeves, purveyor of the excellent ChemTube3D site, contacted me about the transition state (I have commented previously on this aspect of aromatic electrophilic substitution). ChemTube3D recruits undergraduates to add new entries; Blue Jenkins is one such adding a section on dyes.

The mechanism can be rate limiting either in the initial electrophilic attack (black arrows) or in the subsequent proton removal (red arrows using an intermolecular base such as chloride anion).[2].‡ The product is normally assumed to be the trans-diazo compound rather than cis. This distribution is certainly true in the crystal structure database (below, although some examples of cis are known, including azobenzene itself). Would this distribution be reflected in the transition states? Initial attempts by the ChemTube3D team had resulted only in a cis-transition state being located, and they asked me to check this out.
ωB97XD/6-311G(d,p)/SCRF=water calculations using phenyl diazonium chloride (I do like my counter-ions) coupling to benzene resulted in location of both cis[3] and trans[4] transition states, the former being the lower by 1.0 kcal/mol in free energy (this might well be due to the dispersion stabilisation from π-π stacking).† The IRC for the cis is shown below.[5]
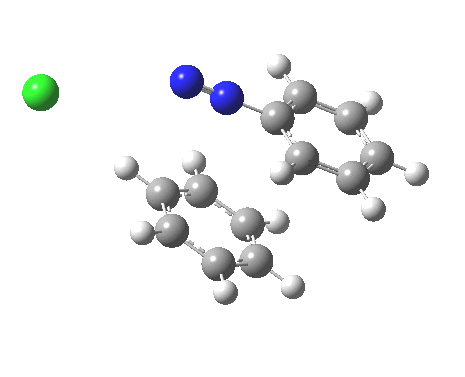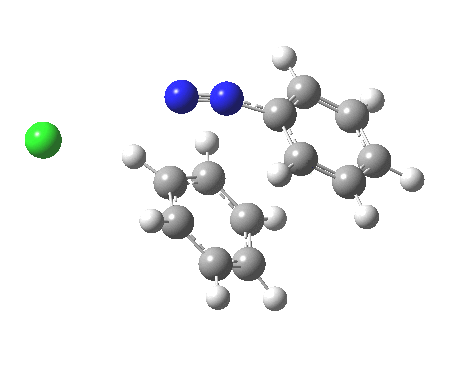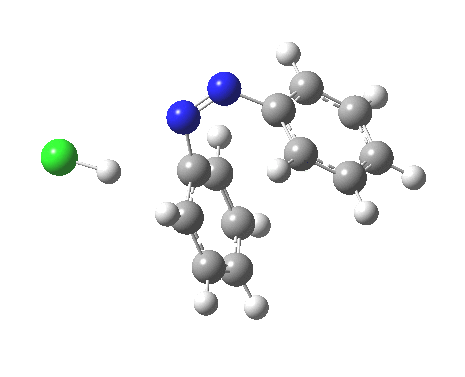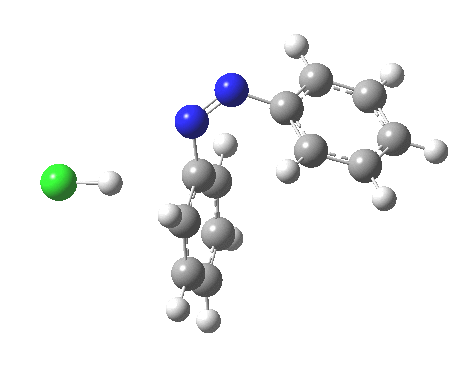


You can see that the entire process is concerted. The Wheland intermediate normally invoked as part of the mechanism of aromatic electrophilic substitution is not a proper intermediate but a hidden one for the reaction with X=Y=H. The reaction coordinate has a flat top, and that passage along this part represents the hidden Wheland. The reaction barrier is high however, and it is certainly observed that only activated arenes (phenols, anilines, X,Y=OH, NH2) actually couple with diazonium cations. For these, the hidden intermediate is stabilized by the substituent, and no doubt emerges as a real intermediate.
For my thesis work, I studied[2] diazo-coupling of indoles. I might have a go at returning to that work, to see if calculations can replicate my finding, that for unhindered indoles proton removal from the Wheland intermediate is fast, but add a few t-butyl hindering groups and it becomes slow.
PS. Here is the IRC for the formation of trans-diazobenzene.[6]
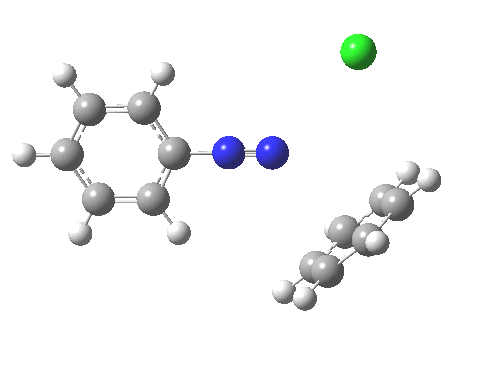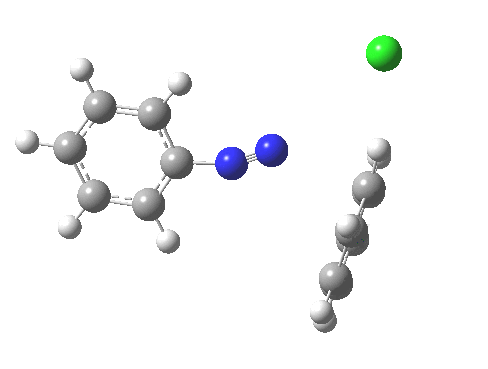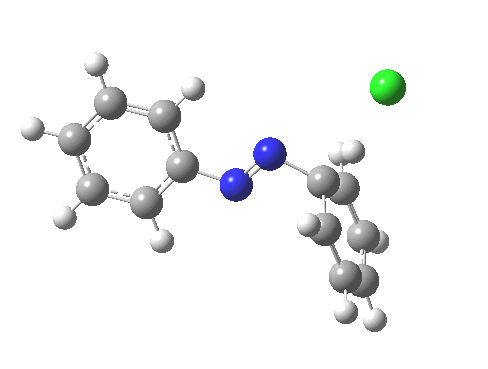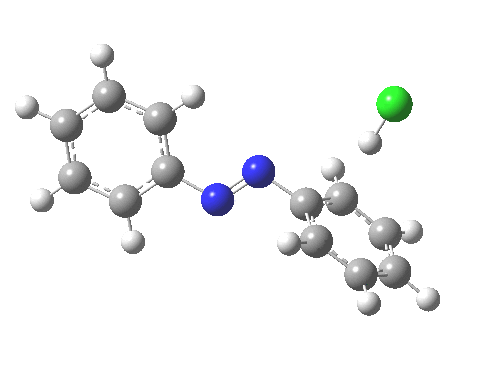
‡Such diazo compounds make up a significant proportion of the 50 or so real molecules I have personally added to the collection of 84 million or so thus far identified.
†Working with ions has one statistical problem that covalent systems do not have; where to geometrically place the counter-ion. One should really stochastically explore reasonable locations before concluding the likely location of the globally lowest energy pose.
Author
References
- S.B. Hanna, C. Jermini, H. Loewenschuss, and H. Zollinger, "Indices of transition state symmetry in proton-transfer reactions. Kinetic isotope effects and Bronested's .beta. in base-catalyzed diazo-coupling reactions", Journal of the American Chemical Society, vol. 96, pp. 7222-7228, 1974. https://doi.org/10.1021/ja00830a009
- B.C. Challis, and H.S. Rzepa, "The mechanism of diazo-coupling to indoles and the effect of steric hindrance on the rate-limiting step", Journal of the Chemical Society, Perkin Transactions 2, pp. 1209, 1975. https://doi.org/10.1039/p29750001209
- H.S. Rzepa, "Gaussian Job Archive for C12H11ClN2", 2014. https://doi.org/10.6084/m9.figshare.956138
- H.S. Rzepa, "Gaussian Job Archive for C12H11ClN2", 2014. https://doi.org/10.6084/m9.figshare.956139
- H.S. Rzepa, "Gaussian Job Archive for C12H11ClN2", 2014. https://doi.org/10.6084/m9.figshare.956209
- H.S. Rzepa, "Gaussian Job Archive for C12H11ClN2", 2014. https://doi.org/10.6084/m9.figshare.956213
Tags: covalent systems, first professor, free energy, Imperial College, lowest energy pose, Nick Greeves, professor of chemistry
Henry, Boris Galabov, Fritz Schaefer, and our collaborators have been computing the mechanisms of various aromatic electrophilc substitution reactions and have noted the LACK of Wheland intermediates, when the counter ions are included.
Instead, the reactions are concerted or go via competing addition-elimination routes. Your present example shows that the arenium ion is not a “proper intermediate,” but only a “hidden” one.
Paul,
The tradition has long been to study eg reactions of cations without a counter-ion, presumed passive. I have felt for a few years this was not correct. Witness thus my discussions with Dan Singleton regarding his molecular dynamics simulation of the addition of a thiolate anion to a chlorobutenone. His simulation did not include the sodium cation as counter-ion. It is entirely possible that including it would make no difference to the outcome, but one does not know this until one tries it.
Of course, introducing a coiner-ion does cause problems. Where to put it for example (there is no directional covalent bond to guide us). Has one explored all the likely locations for it? Does it move during reaction? Should one include explicit solvent molecules surrounding it?
But I remain convinced that one should build what I call a “bottlable” model (one that could exist inside a specimen tube) wherever possible.