Symbiosis between computation and experiment is increasingly evident in pedagogic journals such as J. Chemical Education. Thus an example of original laboratory experiments[1],[2] that later became twinned with a computational counterpart.[3] So when I spotted this recent lab experiment[4] I felt another twinning approaching.
The reaction under consideration is that between dec-2-enal and 2,4-dinitrobenzyl chloride as catalysed by an α,α-diphenylprolinol trimethylsilyl ester with addition of further base (di-isopropylamine?). The proposed mechanism can be seen in figure 7‡ of the journal article[4] and also scheme 2 of an earlier article.[5] The following is my interpretation of their published mechanism (the compound numbering is the same as in Figure 7).
- The initiating step is the condensation between the alkyl enal (1) and the prolinol derivative (3), with elimination of water and the formation of a positive iminium cation (5). One might wonder at this stage what the counter ion to this cation is.
- 5 then reacts with 2,4-dinitrobenzyl chloride (2) with apparent elimination of HCl to form 6. This corresponds to 1,4-Michael addition to 5 with the formation of the first new C-C bond and the creation of two new stereogenic centres.
- 6 then cyclises to form a second new C-C bond and a third new stereogenic centre as in 7.
- 7 is then hydrolysed to give the final product 4.
A total of three (starred) stereogenic centres are therefore created in 4, implying 23 = 8 steroisomers, arranged as four diastereomers and their enantiomers. A computational mechanistic analysis might strive to cast light on the following questions.
- Is the sequence shown in figure 7 reasonable? If not can a more reasonable cycle be constructed that has energetics corresponding to a facile reaction at 0°C?
- What are the predicted relative yields of the four possible diastereomeric products and do they match those observed?
- If R=α,α-diphenylprolinol trimethylsilyl ester, then this fourth chiral centre increases the total number of stereoisomers to 16, arranged in eight pairs of diastereomers. Does this result in the diastereomers of 4 forming with an excess of one enantiomer over the other (an ee ≠ 0)?
This post addresses just the first question (R=R’=H, R”=isopropylamine) leaving the other two questions for later analysis.

My analysis (figure above)♥ of the mechanism, as cast for computational analysis†, differs in various details from Figure 7/Scheme 2 of the published articles.[4],[5]
- The issue of defining a counterion to 5 is solved by in fact starting the cycle with proton abstraction from 2 by di-isopropylamine♦ to form a benzylic anion, as stabilized by the 2,4-dinitro groups and with the positive counter-ion being the protonated amine base.
- The next step is reaction between 1 and 3 to form an aminol 10, a tetrahedral intermediate.
- To remove water from this to form an iminium cation 5, one has to protonate the hydroxy group and this can now be done using the cationic ammonium species formed in step 5 above.
- The benzylic anion can now react with the iminium cation to form the first C-C bond and the first two stereocentres via 1,4-Michael addition to form 6
- The species 6 can now eliminate chloride anion to form the cyclopropyl iminium cation/anion pair 7, generating the 3rd stereogenic centre.
- Hydrolysis forms the product 4 and returns the system to the starting point in the catalytic cycle.
- Also included is whether an alternative mechanism is viable, involving elimination of Cl– from 8 to form a “carbene”, which could then potentially add to the alkene in 1.
|
Species (transition state) FAIR Data DOI |
ΔG273.15, Hartree |
Structure |
|---|---|---|
| Reactants | -1837.174744♣ (0.0) | 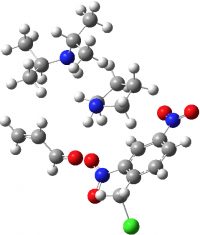 |
| TS1 | -1837.150502 (15.2) | 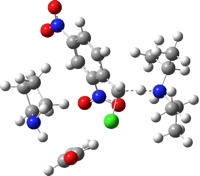 |
| TS2 | -1837.154923 (12.4) | 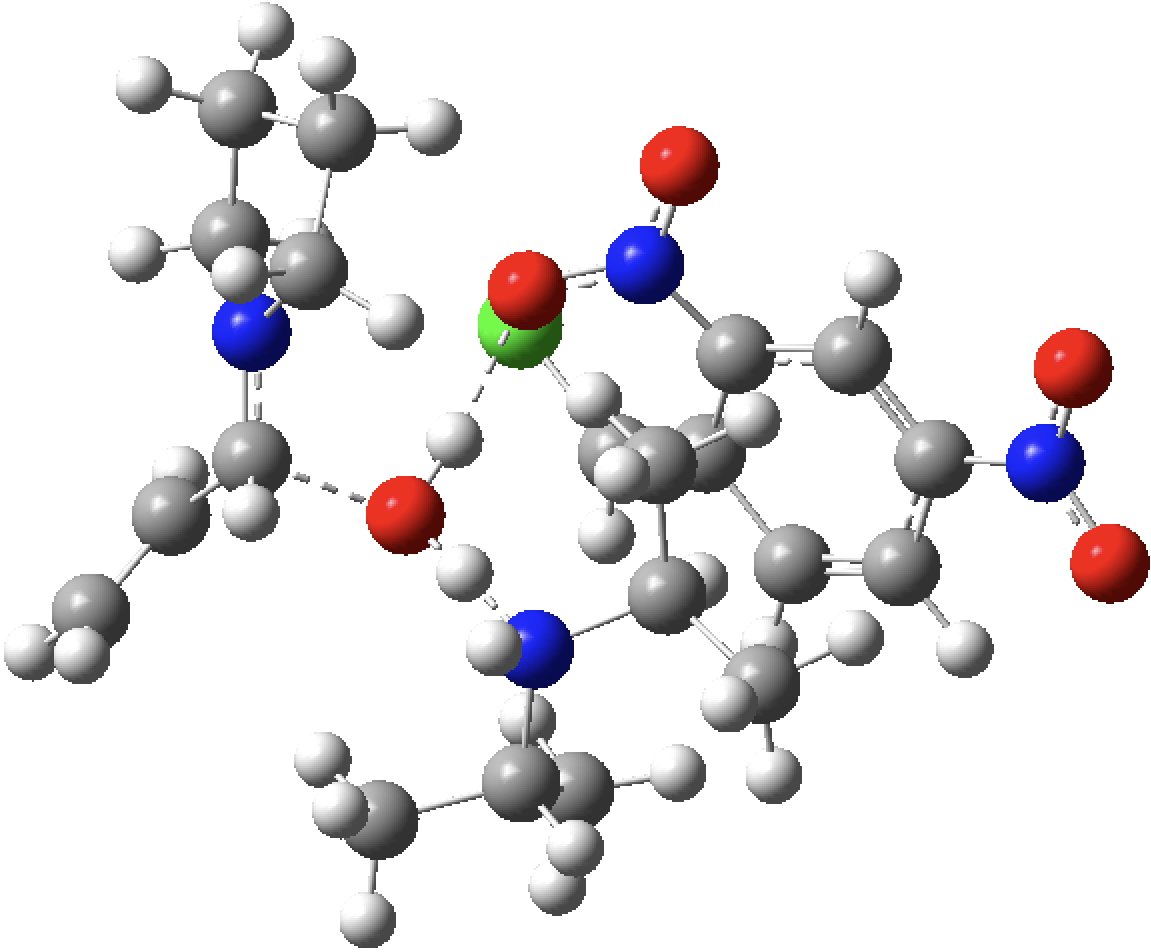 |
| TS3 | -1837.147927 (16.8) | 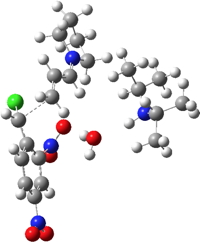 |
| TS4 | -1837.175723 (-0.6) | 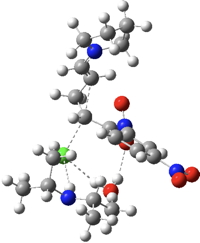 |
| TS5 | -1837.101534 (45.9) | 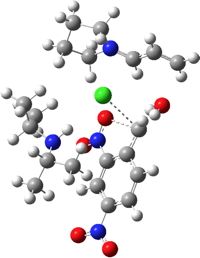 |
The (relative) free energies of the transition states at the B3LYP+GD3BJ/6-311G(d,p)/SCRF=chloroform level shown in the table above (click on the thumbnail images to show the 3D model of each transition state) reveal that the highest point corresponds to TS3, a C-C bond forming reaction. This is noteworthy because it constitutes the reaction between an ion-pair, albeit ions which are both heavily stabilized by delocalisation. Since the reaction is known to proceed over 3 hours at 0°C, the activation barrier of 16.8 kcal/mol is also entirely reasonable. TS5, the putative formation of a carbene from the benzyl chloride, has a very high barrier and in fact cyclises to form 9. This pathway can therefore be safely ignored.
The next stage would be to investigate the stereochemical implications of this mechanism (atoms in 4 marked with a *) using the actual substituents for R and R’. Because the mechanism includes ion-pairs throughout, this does actually present some tricky issues. Unlike molecules with covalent bonds, where the shapes are relatively easy to predict, ion-pairs are more flexible and can often adopt a variety of poses, the relative energy of which is frequently determined simply by the magnitudes of their dipole moments.[6] If I manage to sort this out, I will report back here.
‡I would love to show you figure 7 here, but the publisher asserts that I would need to pay them $87.75 to do so and so you will have to acquire the article yourself to see it.
†Various guiding rules include constructing the entire catalytic cycle using exactly the same number of atoms so that the cycle can show only relative (free) energies and using neutral ion-pair models rather than just charged species alone.
♥Almost all the chemical diagrams on this blog for some ten years now have been in SVG (scalable vector graphics) format. Most modern web browsers for a number of years now have had excellent support for SVG. Until recently SVG could not be generated directly from a drawing program such as e.g. ChemDraw. Instead I saved as EPS (encapsulated postscript) and then used a program called Scribus to convert to SVG. In fact with Chemdraw V18.0, the direct conversion to SVG seems to be working very well, including honoring color maps. To scale up a diagram, click on it to open a new browser window containing only it and then use the browser zoom-in control to magnify it. Unlike e.g. a pixel image, SVG images magnify/scale correctly.
♣This relates to metadata as described in this post in performing a global search of any species matching this Gibbs Energy.
♦If the mechanism is set up without any base, then proton abstraction must occur directly from the benzyl chloride. Under these circumstances, the barrier for proton removal is 27.5 kcal/mol, whilst that for C-C bond formation is only 13.6.
Author
References
- A. Burke, P. Dillon, K. Martin, and T.W. Hanks, "Catalytic Asymmetric Epoxidation Using a Fructose-Derived Catalyst", Journal of Chemical Education, vol. 77, pp. 271, 2000. https://doi.org/10.1021/ed077p271
- J. Hanson, "Synthesis and Use of Jacobsen's Catalyst: Enantioselective Epoxidation in the Introductory Organic Laboratory", Journal of Chemical Education, vol. 78, pp. 1266, 2001. https://doi.org/10.1021/ed078p1266
- K.K.(. Hii, H.S. Rzepa, and E.H. Smith, "Asymmetric Epoxidation: A Twinned Laboratory and Molecular Modeling Experiment for Upper-Level Organic Chemistry Students", Journal of Chemical Education, vol. 92, pp. 1385-1389, 2015. https://doi.org/10.1021/ed500398e
- M. Meazza, A. Kowalczuk, S. Watkins, S. Holland, T.A. Logothetis, and R. Rios, "Organocatalytic Cyclopropanation of (<i>E</i>)-Dec-2-enal: Synthesis, Spectral Analysis and Mechanistic Understanding", Journal of Chemical Education, vol. 95, pp. 1832-1839, 2018. https://doi.org/10.1021/acs.jchemed.7b00566
- M. Meazza, M. Ashe, H.Y. Shin, H.S. Yang, A. Mazzanti, J.W. Yang, and R. Rios, "Enantioselective Organocatalytic Cyclopropanation of Enals Using Benzyl Chlorides", The Journal of Organic Chemistry, vol. 81, pp. 3488-3500, 2016. https://doi.org/10.1021/acs.joc.5b02801
- J. Clarke, K.J. Bonney, M. Yaqoob, S. Solanki, H.S. Rzepa, A.J.P. White, D.S. Millan, and D.C. Braddock, "Epimeric Face-Selective Oxidations and Diastereodivergent Transannular Oxonium Ion Formation Fragmentations: Computational Modeling and Total Syntheses of 12-Epoxyobtusallene IV, 12-Epoxyobtusallene II, Obtusallene X, Marilzabicycloallene C, and Marilzabicycloallene D", The Journal of Organic Chemistry, vol. 81, pp. 9539-9552, 2016. https://doi.org/10.1021/acs.joc.6b02008
Tags: Ammonium, Benzyl group, Cations, chemical diagrams, Chemistry, condensation, final product, Functional groups, Iminium, Methyl group, Name reactions, Organic chemistry, possible diastereomeric products, relative energy, Vector Graphics, web browsers
Here is a thought about the stereochemistry. The three new chiral centres are created during two different transition states, only one of which is rate determining (TS3, the first, rather than TS4, the second). So any stereospecificity induced in the third stereocentre cannot be due to the properties of TS3. Could it be due to reaction dynamics instead?
Hi Henry,
1. The base which the authors used was DIPEA = diisopropyl-ethylamine, or “Hünigs base”, as the Germans like to call it. It is thus tertiary, but the basicity may be similar.
2. The loss of Cl- in the authors original scheme on the way from 6 to 7 is probably a mistake. Alternatively, they might have invoked chloride as counter-ion for iminium ion 6, which could make sense after a few turnovers into the catalysis, with chloride accumulating. With 20 mol-% of catalyst, the number of turnovers is limited, though…
3. Would the basicity of the OH-group in hemiaminal 10 be sufficient to deprotonate the benzylic chloride? In that sense, the external base might only serve as acid quencher to keep the acidity level low. Since K2CO3 was also supporting the reaction (less effectively), the role of the base could be passive.
4. A reason for my point 3 is the following: Aminal formation from aldehyde and secondary amine (pyrrolidine in particular) is mediated in the presence of solid K2CO3! (Mannich, Chem. Ber. 1936, 69, 2106). Not sure what is the role of K2CO3 (Mannich thinks it removes water), but it is notable for what appears to be a base-catalyzed imination (or the like), which are usually though of as acid-catalyzed. So the tendency of OH in hemiaminals to leave as OH- appears to be relatively strong. Maybe that OH-group is fairly basic, or becomes so as the C–OH bond elongates.
Let me comment on each of your four points separately.
1. In the text of the article, the authors refer to the base DIPEA, but apparently equating it to di-isopropylamine in their text. I did contact them for definitive clarification, and although they acknowledged my query, I do not yet have a full answer. The SI does indicate more clearly it was DIPEA.
2. The diagram in the J. Chem. Ed. journal is a direct reproduction from the original article in J. Org. Chem. Yes, the location for elimination of HCl does seem an error. If the counter-ion for the iminium cation formation is not Cl(-), it could only be hydroxide anion, which is too poor a leaving group to act in this manner. If it is an error, then both articles should be corrected.
When writing for an audience of students, one should I think strive for clarity whenever possible. And from a pedagogic point of view, I would also like to see more formal “curly arrows” in the scheme. The one arrow shown is probably not curly, but more purely schematic?
3. I did actually calculate a TS for the OH group in the hemiaminal directly deprotonating the benzyl chloride. The activation free energy barrier was 27.5 kcal/mol(DOI: 10.14469/hpc/4633) which provides a good explanation for why additional base is added to promote the reaction.
4. The reported reaction occurs rapidly at 0°C, which suggests very rapid deprotonations and C-C bond formations. Certainly the role of a solid reagent is far more inscrutable than a soluble one!
“the highest point corresponds to TS3, a C-C bond forming reaction. This is noteworthy because it constitutes the reaction between an ion-pair…”
This reminds me of something I read a long time ago: In 1998, J. Dale (Oslo Univ.) wrote about “Inadequacies of the SN1 Mechanism”. His point was basically that free carbenium ions were too reactive as to exist in nucleophilic solvents (water) as solvated species, since if water was present in the vicinity of a carbenium ion for solvating it, there would be no reason for the water to stop “… and proceed the tiny remaining distance and form a stable chemical bond.” Thus he argued that the SN1 mechanism (in water/alcohols?) was not realistic and that racemization was the result of double inversion with water as nucleophile.
Not sure if he is (generally) correct; with more stabilized carbenium ions (e.g. iminium, but als trityl etc) the attack of nucleophiles can be slow (as your above calculation shows even with opposite charges), and the presence of solvated carbenium ions seems quite sensible. Also Mayr’s “carbocation watching” experiments (with stabilized benzhydrylium ions) leave little doubt about the presence of “free” carbenium ions and SN1 type mechanisms.
Your comments about the instability of a carbenium ion in SN1 reaction conditions are reflected in a reaction profile of t-butyl chloride with 16 water molecules that I showed a few years back; https://www.ch.imperial.ac.uk/rzepa/blog/?p=5228
The IRC shows the carbenium ion to be a hidden intermediate (at around IRC 7 below), which collapses by inverting attack of a water molecule opposite to the direction of the leaving chloride. This suggests that the classical transition state may not be a good reflection on the dynamic picture. No doubt various water trajectories can succeed, and so the rate is probably controlled by many trajectories, not all of which pass through the geometric region of the transition state.
Another ion-pair that can exhibit a barrier to reaction is hydronium hydroxide. Unlikely though it might seem, there are some arrangements of this ion-pair that show a barrier to proton transfer to form water; https://www.ch.imperial.ac.uk/rzepa/blog/?p=16118
Again this is a dynamic problem, and most arrangements of hydronium hydroxide with solvating water molecules probably undergo proton transfer with no barrier.