Posts Tagged ‘potential energy surface’
Thursday, April 4th, 2019
Previously, I explored the Graham reaction to form a diazirine. The second phase of the reaction involved an Sn2′ displacement of N-Cl forming C-Cl. Here I ask how facile the simpler displacement of C-Cl by another chlorine might be and whether the mechanism is Sn2 or the alternative Sn1. The reason for posing this question is that as an Sn1 reaction, simply ionizing off the chlorine to form a diazacyclopropenium cation might be a very easy process. Why? Because the resulting cation is analogous to the cyclopropenium cation, famously proposed by Breslow as the first example of a 4n+2 aromatic ring for which the value of n is zero and not 1 as for benzene.[1] Another example of a famous “Sn1” reaction is the solvolysis of t-butyl chloride to form the very stable tertiary carbocation and chloride anion (except in fact that it is not an Sn1 reaction but an Sn2 one!)
The reason for posing this question is that as an Sn1 reaction, simply ionizing off the chlorine to form a diazacyclopropenium cation might be a very easy process. Why? Because the resulting cation is analogous to the cyclopropenium cation, famously proposed by Breslow as the first example of a 4n+2 aromatic ring for which the value of n is zero and not 1 as for benzene.[1] Another example of a famous “Sn1” reaction is the solvolysis of t-butyl chloride to form the very stable tertiary carbocation and chloride anion (except in fact that it is not an Sn1 reaction but an Sn2 one!)
(more…)
References
- R. Breslow, "SYNTHESIS OF THE s-TRIPHENYLCYCLOPROPENYL CATION", Journal of the American Chemical Society, vol. 79, pp. 5318-5318, 1957. https://doi.org/10.1021/ja01576a067
Tags:animation, Carbenium ion, Cations, Chemical elements, chemical reaction, Chemistry, Chlorine, computational chemistry, Cyclopropenium ion, Diazirine, energy, energy profile, free energy, Halogens, Natural sciences, Nucleophilic aromatic substitution, Oxidizing agents, Physical sciences, potential energy surface, SN1 reaction, Substitution reactions
Posted in reaction mechanism | No Comments »
Friday, December 21st, 2018
Five years back, I speculated about the mechanism of the epoxidation of ethene by a peracid, concluding that kinetic isotope effects provided interesting evidence that this mechanism is highly asynchronous and involves a so-called “hidden intermediate”. Here I revisit this reaction in which a small change is applied to the atoms involved.
(more…)
Tags:Chemical kinetics, chemical reaction, Chemistry, Deuterium, Isotope effect, Kinetic isotope effect, Natural sciences, Organic chemistry, overall activation energy, pericyclic reaction, Physical organic chemistry, Physical sciences, potential energy surface, Rearrangement reactions
Posted in Interesting chemistry | 5 Comments »
Sunday, December 17th, 2017
Alkalides are anionic alkali compounds containing e.g. sodide (Na–), kalide (K–), rubidide (Rb–) or caeside (Cs–). Around 90 examples can be found in the Cambridge structure database (see DOI: 10.14469/hpc/3453 for the search query and results). So what about the ammonium analogue, ammonide (NH4–)? A quick search of Scifinder drew a blank! So here I take a look at this intriguingly simple little molecule.‡
(more…)
Tags:Alkalide, Ammonium, Anions, Atomic physics, Chemistry, electron gas, energy, free-electron gas, Jahn-Teller, kinetic energy density, Matter, Nitrogen, potential energy surface, search query
Posted in Hypervalency | 2 Comments »
Sunday, October 1st, 2017
I noted in my WATOC conference report a presentation describing the use of calculated reaction barriers (and derived rate constants) as mechanistic reality checks. Computations, it was claimed, have now reached a level of accuracy whereby a barrier calculated as being 6 kcal/mol too high can start ringing mechanistic alarm bells. So when I came across this article[1] in which calculated barriers for a dyotropic ring expansion observed under mild conditions in dichloromethane as solvent were used to make mechanistic inferences, I decided to explore the mechanism a bit further.
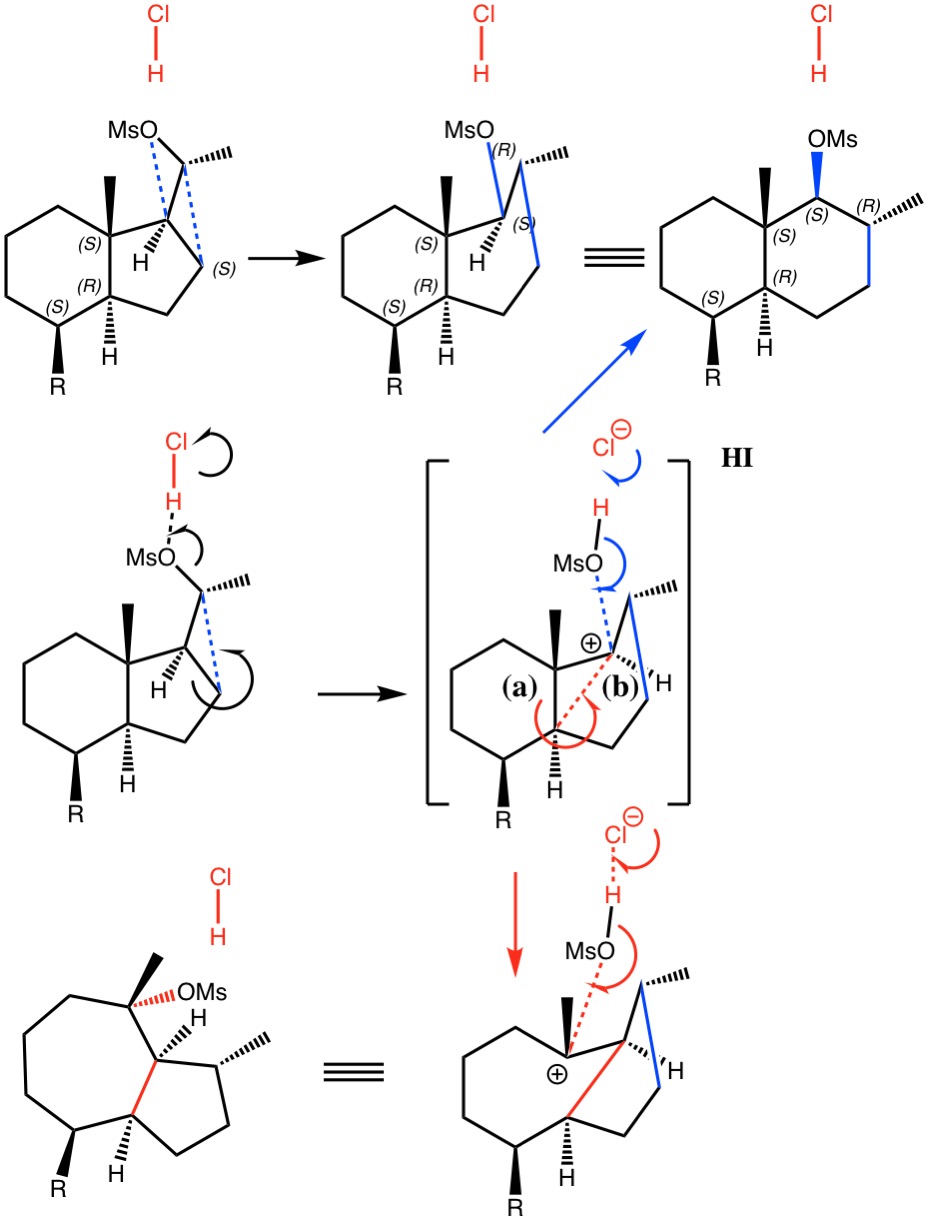
(more…)
References
- H. Santalla, O.N. Faza, G. Gómez, Y. Fall, and C. Silva López, "From Hydrindane to Decalin: A Mild Transformation through a Dyotropic Ring Expansion", Organic Letters, vol. 19, pp. 3648-3651, 2017. https://doi.org/10.1021/acs.orglett.7b01621
Tags:animation, bicyclic ring product, energy derivative gradient norm, energy profile, final non-ionic product, Organic chemistry, possible products, potential energy surface, realistic model for the reaction
Posted in pericyclic, reaction mechanism | 3 Comments »
Sunday, April 9th, 2017
Both the cyclopropenium cation and the cyclopentadienide anion are well-known 4n+2-type aromatic ions, but could the two together form an ion-pair?

(more…)
Tags:Anions, Aromatization, Cation–pi interaction, Chemistry, Cyclopentadienyl anion, Ion, Ion association, potential energy surface, Simple aromatic rings
Posted in crystal_structure_mining, Interesting chemistry | 6 Comments »
Saturday, April 1st, 2017
In a comment appended to an earlier post, I mused about the magnitude of the force constant relating to the interconversion between a classical and a non-classical structure for the norbornyl cation. Most calculations indicate the force constant for an “isolated” symmetrical cation is +ve, which means it is a true minimum and not a transition state for a [1,2] shift. The latter would have been required if the species equilibrated between two classical carbocations. I then pondered what might happen to both the magnitude and the sign of this force constant if various layers of solvation and eventually a counter-ion were to be applied to the molecule, so that a bridge of sorts between the different states of solid crystals, superacid and aqueous solutions might be built.
(more…)
Tags:Carbocations, chemical bonding, Chemistry, constant matrix/search direction, continuum model for water, gas phase, Paul Schleyer, Physical organic chemistry, potential energy surface, Reactive intermediates, superacid and aqueous solutions
Posted in crystal_structure_mining, Interesting chemistry, reaction mechanism | 7 Comments »
Monday, October 31st, 2016
Is asking a question such as “what is the smallest angle subtended at a chain of three connected 4-coordinate carbon atoms” just seeking another chemical record, or could it unearth interesting chemistry?
(more…)
Tags:animation, Bicyclic molecule, chemical record, Chemistry, City: Cambridge, Cycloalkane, Cyclopropanes, Java, Molecular geometry, Organic chemistry, potential energy surface, Safari, Web browser, X-ray
Posted in crystal_structure_mining, reaction mechanism | 7 Comments »
Friday, April 10th, 2015
Previously on this blog: modelling the reduction of cinnamaldehyde using one molecule of lithal shows easy reduction of the carbonyl but a high barrier at the next stage, the reduction of the double bond. Here is a quantum energetic exploration of what might happen when a second LAH is added to the brew (the usual ωB97XD/6-311+G(d,p)/SCRF=diethyl ether).
(more…)
Tags:computed free energy barrier, energy, energy surface, final product, flat energy potential, free energy, lower energy pathways, metal exchange, pence, potential energy surface, reduction, Yes
Posted in reaction mechanism | No Comments »
Thursday, October 9th, 2014
This second report highlights two “themes”, or common ideas that seem to emerge spontaneously from diversely different talks. Most conferences do have them.
(more…)
Tags:Complex Biological Systems, condensation, gas-phase molecular species, metal surface catalysis, molecular systems, non-crystalline systems, organic chemist, organometallic systems, potential energy surface, representative, Stefan Grimme, Thus Emily Carter
Posted in Interesting chemistry, WATOC reports | 1 Comment »
Sunday, February 16th, 2014
The potential energy surface for a molecule tells us about how it might react. These surfaces have been charted for thousands of reactions using quantum mechanics, and their basic features are thought to be well understood. Coming across an entirely new feature is rare. So what do you make of the following?
(more…)
Tags:potential energy surface
Posted in pericyclic, reaction mechanism | 11 Comments »