George Olah passed away on March 8th. He was part of the generation of scientists in the post-war 1950s who had access to chemical instrumentation that truly revolutionised chemistry. In particular he showed how the then newly available NMR spectroscopy illuminated structures of cations in solvents such “Magic acid“. The obituaries will probably mention his famous “feud” with H. C. Brown over the structure of the norbornyl cation (X=CH2+), implicated in the mechanism of many a solvolysis reaction that characterised the golden period of physical organic chemistry just before and after WWII.

The dispute between Olah and Brown was not played on a pitch using quite the same goal posts. Olah did much of his work in magic acid and Brown did his in aqueous solutions. I was involved in a tiny way when the discussion about the precise character of the norbornyl cation was reaching its peak in the mid 1970s. At the time, I was working with Michael Dewar, who was himself not shy in joining in the fun and sometimes very acrimonious disputes at conferences. We contributed by calculating the so-called core-electron carbon ESCA spectrum.[1] History records that we came down on the wrong side,‡ by suggesting that this form of spectroscopy supported Brown rather than Winstein/Olah on the basis of a 6:1 spectral deconvolution (classical) rather than 5:2 (non-classical). More recently of course the crystal structure of the parent cation itself has been shown to be non-classical[2] (there are other crystal structures which differ in respect to having one or more additional methyl groups[3]). For a 3D model of norbornyl cation, see DOI: 10.5517/CCZ21LN. This still leaves the issue (very slightly) open for the structure of the solvated cation when formed in water!
When I started to teach a course in molecular modelling, I touched briefly on how modelling could contribute and whilst updating the notes in the 1990s, wondered why the boron analogue had never been so studied (X=BH2). Unlike the crystallographically difficult norbornyl ion-pair, the iso-electronic boron species would be neutral and not need a counter-ion. Perhaps it might be a more manageable molecule? Checking the Cambridge structural database, such a species has never been reported!† So here as my homage to Olah, I report its calculated structure (b2plypd3/Def2-TZVPP, DOI: 10.14469/hpc/2236).
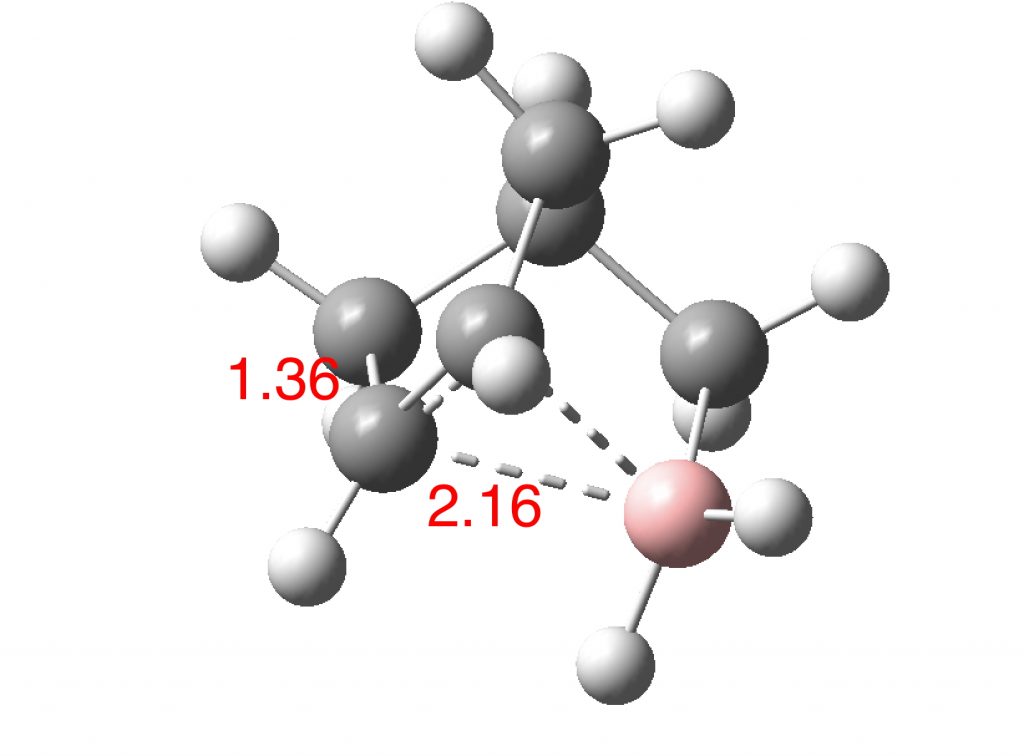
The norbornyl cation has symmetrical C-C bridging distances of ~1.80±0.02Å and a basal C-C distance of ~1.39±0.02Å. The calculated values for the boron equivalent are 2.16Å and 1.36Å respectively, with all positive force constants. B-C bonds are normally 1.66-1.72Å, significantly longer than C-C bonds, which makes the longer B-C lengths in this example unsurprising. More interestingly, the species has one vibrational normal mode (ν 203 cm-1) which corresponds to the [1,2] shift of the BH2 group across the basal C-C. For a classical species, this vibrational motion would correspond to a transition state (an imaginary vibration) but for a non-classical species it is of course real. In this sense it is analogous to the so-called real Kekulé mode in non-classical benzene, which “equilibrates” the two classical Kekulé structures. The corresponding calculated vibration for the norbornyl cation itself is ν 194 cm-1 (DOI: 10.14469/hpc/2238).
Of course, the entire controversy over the structure of this species is littered with comparisons between not quite similar systems, differing in a methyl group more or less. So morphing a C+ to a B might be seen as quite a large change. But perhaps if it had been crystallised in say the 1960s, would the subsequent debates have taken a different turn?
‡ We were also wrong about the symmetry of the Diels-Alder cyclisation, which is nowadays accepted to be synchronous rather than asynchronous for simple Diels-Alder reactions. But that is another story.
†GAXLIA is perhaps the closest analogue.[4],
Author
References
- M.J.S. Dewar, R.C. Haddon, A. Komornicki, and H. Rzepa, "Ground states of molecules. 34. MINDO/3 calculations for nonclassical ions", Journal of the American Chemical Society, vol. 99, pp. 377-385, 1977. https://doi.org/10.1021/ja00444a012
- https://doi.org/
- T. Laube, "Redetermination of the Crystal Structure of the 1,2,4,7‐<i>anti</i>‐tetramethylbicyclo[2.2.1]heptan‐2‐yl cation at 110 K", Helvetica Chimica Acta, vol. 77, pp. 943-956, 1994. https://doi.org/10.1002/hlca.19940770407
- P.J. Fagan, E.G. Burns, and J.C. Calabrese, "Synthesis of boroles and their use in low-temperature Diels-Alder reactions with unactivated alkenes", Journal of the American Chemical Society, vol. 110, pp. 2979-2981, 1988. https://doi.org/10.1021/ja00217a053
Tags: 2-Norbornyl cation, aqueous solutions, Chemical bond, chemical instrumentation, Chemistry, George Andrew Olah, George Olah, Ion association, Magic acid, Michael Dewar, Molecule, Nature, Physical organic chemistry, Reactive intermediates, spectroscopy
Better to make one’s best guess and be proven wrong, than to sit on the sidelines and never try to discover anything new!
A semi-related question — I’m reading Aaron Ihde’s History of Chemistry (https://www.amazon.com/Development-Modern-Chemistry-Dover-Books/dp/0486642356) right now and really enjoying it. It was written fifty years ago, though, and so it captures little to none of the “post-war … chemical instrumentation that truly revolutionised chemistry.”
Is there any literature you can recommend that covers well the history of chemistry, especially instrumental, over the last half of the 20th century?
I looked briefly at Brock’s 1992 “The Chemical Tree” (https://www.amazon.com/Chemical-Tree-History-Chemistry-2000-06-17/dp/B01N6KSKAI), but it doesn’t seem to have much of a history of instrumental topics (or of organometallic chemistry, for that matter).
Brian,
Regarding histories of chemical instrumentation, perhaps Peter Morris’ very recent book The Matter Factory: A History of the Chemistry Laboratory might cover some of this aspect?
It is also worth noting that great structural breakthroughs: were probably made using the polarimeter as a spectrometer, which was of course available during most of the 19th century. Indeed, Meerwein’s famous proposal of carbocations themselves in the early 1920s arose with the help of this instrument.
He was indeed a great scientist though I knew him just by his papers and biography. Indeed sending this post as a tribute to his achievements was a good idea.
Very sad to read the passing of George Olah.
I thought it might be interesting to summarise the essential difference between a classical and a non-classical carbocation, as generalised to other examples in chemistry. If anyone can think of further systems, please tell!
I noted above that the [1,2] shifting mode for the charged norbornyl cation was 194 cm-1. Applying a solvation field for water (DOI: 10.14469/hpc/2243) increases this slightly to 203cm-1. This suggests that aqueous solvation may slightly increase the non-classical character of this species, and hence that the species is just as likely to retain this character in water. Of course, the effect of explicit hydrogen bonding from the water on this character still remains to be established.
Paul Schleyer gave a seminar in summer 2014 at LMU in Muenchen on the nonclassical 2-norbornyl cation, since he was also very much engaged in this topic together with Olah. He pointed out that the final solution was obtained by his recent paper “Crystal Structure Determination of the 2-Norbornyl Cation” in Science 2013, 341, 62-64.
We have had some discussion about what Brown would say about the last result?
May be he would say: that is not the answer. My question is what is the reason for the large exo-endo rate and product ratio.
Dieter,
Yes indeed, the Schleyer article is cited as Ref 2 in the post above. I am sure that Brown would say it may not be the last result, since the structure in superacids and in the crystalline state may not be the same as the ion in hydrogen bonding solvents such as water, where the large exo-endo rates and products are observed. I too had many a discussion with Paul about the nature of the solvolysis reactions, indeed Paul asked me to locate the TS for the ionisation of a number of them, but this was an unfinished project when Paul died (we did manage to publish such TS which included explicit solvent molecules for another system in 2010, DOI: 10.1021/jo100920e) This is why the issue is still open (Brown might have said more than slightly), but I suspect few researchers are interested any more in resolving these outstanding aspects.
I might still repeat the calculation of the structure with the inclusion of a modest number of explicit solvent molecules, and perhaps even with a counter-ion. For cations, C-H… O hydrogen bonding interactions are known to be unusually strong, and who knows it might reveal something unexpected!
It seems to me that the long distances between the 3 carbons in Schleyer’s paper clinches the fact that the bonds are not classical — The bond lengths between the 3 carbons are 1.78 to 1.83 Angstroms — far longer than the classic length of 1.54 Angstroms of a C – C single bond.
What do the practicing organic chemists say? I left the field 55 years ago, but still read about it.
Yes, the assertion above that “the crystal structure of the parent cation itself has been shown to be non-classical” was based on precisely this evidence of bond length. But crystallographic bond lengths CAN sometimes mislead. If the barrier interconverting two forms is very low, then you could have in effect baked in disorder between two equally probable classical species which might make it appear that the bonds are this length. You have to apply highly sophisticated crystallographic techniques to eliminate such models, which I understand was done.
I would refer you to this post where the position of a hydrogen bond in relation to a phenyl ring is discussed. The authors of this neutron diffraction study of ammonium tetraphenylborate wrote “even at 20 K the ammonium ion performs large amplitude motions which allow the N-H vectors to sample the entire face of the aromatic system” although the nominal distance measured from the hydrogen appeared to correspond to it pointing to the ring centroid (the equivalent of the non-classical model).
In the case of the norbornyl cation, the crystallographic evidence was also supported by high level calculations. These show that the force constant for the [1,2] equilibration of two classical structures was positive, thus supporting a non-classical model, rather than negative as would be required for a classical one.
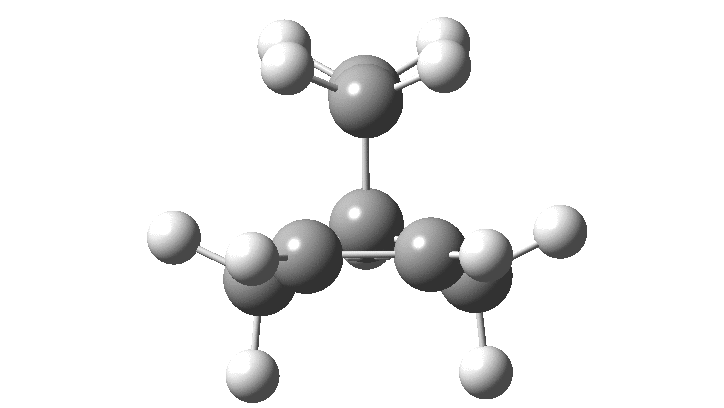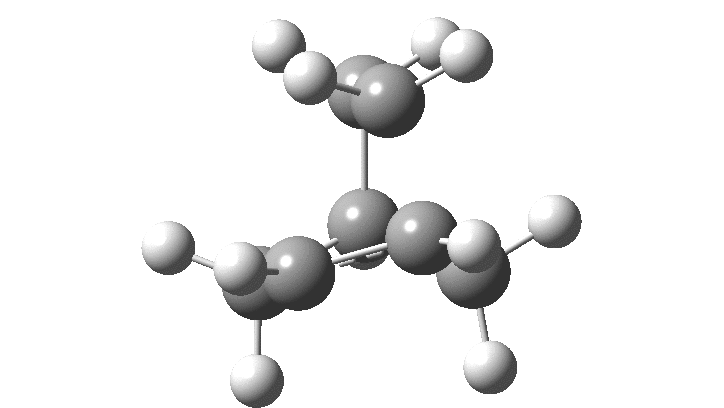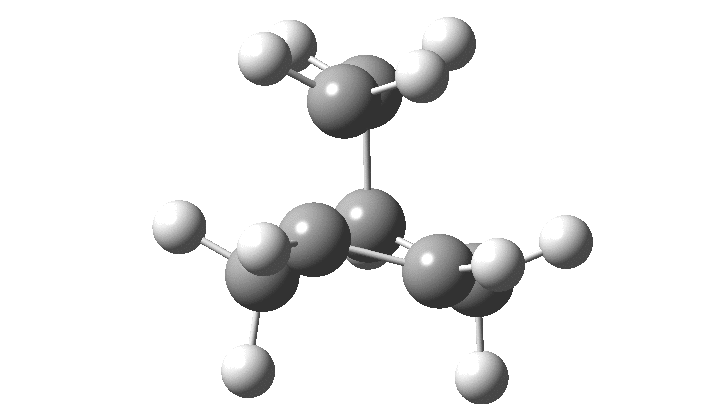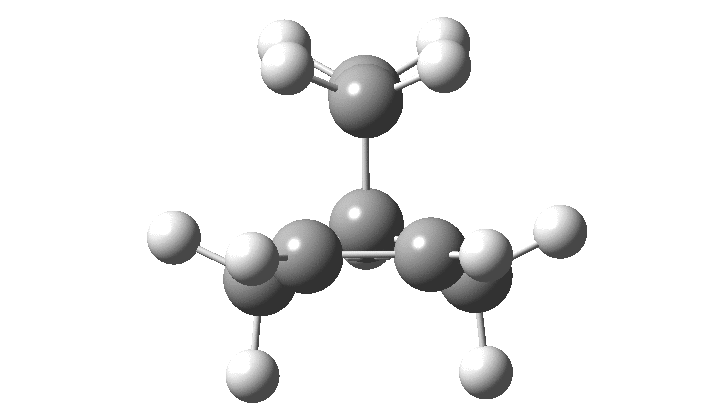
The issue then boils down to whether the sign of this force constant can be influenced by the environment in which the cation finds itself. This would include the counter-ion, the dielectric of the medium it finds itself in and any strong hydrogen bonds that might exist in solution but which are absent in a crystal structure. As I showed in an earlier comment, the continuum dielectric model seems to increase slightly the +ve nature of this force constant, but the other factors have yet to be investigated.
Thanks. Interesting — when you try to do crystallography on an intrinsically disordered protein, you get a blur (which I guess is a sort of an average) but not definite positions of the atoms.
If you’re interested in what it was like to be an undergraduate chemistry major with Schleyer as an advisor nearly 60 years ago please see
https://luysii.wordpress.com/2014/12/15/paul-schleyer-1930-2014-a-remembrance/
and
https://luysii.wordpress.com/2014/12/14/paul-schleyer-1930-2014-r-i-p/
It was a formidable experience
The “blur” that one gets is often associated with anisotropic thermal ellipsoids, in which the elongated axis corresponds to the thermal motions. It is possible to analyse these ellipsoids, which in fact I remember being done for another controversial structure, that of [18] annulene. But even there I recollect the results, whilst strongly suggestive, still allow scope for some uncertainty.
Thanks for the links to the UG experience. We all heard much about these aspects at the symposium in honour of Paul in Erlangen last year from many people.
I add here an even less classical structure recently reported: DOI: 10.5517/CCDC.CSD.CC1M71QM in which an apical carbocationic carbon has a bond length of ~1.7Å to five basal carbons, significantly shorter than for the norbornyl cation itself. It is an excellent example of hexa-coordinated carbon.
I suggested above a boron analogue to norbornyl cation, noting that no examples existed in the Cambridge structure database. At DOI: 10.14469/hpc/2316 I have placed a full search of this database. It reveals structures corresponding to addition to alkenes, but none close to norbornyl.
Did you check the total energy of the bora-analogue of the “classical” norbornyl cation structure? It would be 2-borabicyclo[2.2.1]heptane, presumably a quite stable compound. The non-classical structure (the bora-analogue of the norbornyl-cation) might be more something of a fleeting intermediate (π-complex) in the hydroboration mechanism? My guess is that the if the open-chain version ((cyclopent-3-en-1-ylmethyl)borane) could be prepared, it would suffer intra- or inter-molecular hydroboration.
The “classical” bora-analogue of norbornyl would of necessity need to be zwitterionic, with a negative (formal) separation of charge on boron and a positive charge on the 3-coordinate carbon. Perhaps this alone would weight it strongly towards the bridged “non-classical” form, and as a result make the comparison with norbornyl itself rather unfair. In the latter case, both structures are cationic and no separation of charge is needed.
If a bora-analogue X=BH2– disfavours asymmetry purely on the grounds that this would increase (hetero)charge separation in a neutral molecule, than an isoelectronic aza-form X=NH2+ should do the reverse. It should actively favour de-symmetrisation as a means of achieving (homo)charge separation in a di-cationic molecule.
Below is the optimised structure (B2BLYPD3/Def2-TZVPP/SCRF=water; DOI: 10.14469/hpc/2344). Crucially, this is now characterised as a transition state, with one -ve force constant corresponding to a wavenumber of 391i cm-1 and a free energy of activation of 11.7 kcal/mol.
So the vibrational mode for the [1,2] shift of group X can be tuned anywhere from +ve (B) to -ve (N), with carbon just on the +ve side of the spectrum.