A few years back, I did a post about the Pirkle reagent[1] and the unusual π-facial hydrogen bonding structure[2] it exhibits. For the Pirkle reagent, this bonding manifests as a close contact between the acidic OH hydrogen and the edge of a phenyl ring; the hydrogen bond is off-centre from the middle of the aryl ring. Here I update the topic, with a new search of the CSD (Cambridge structure database), but this time looking at the positional preference of that bond and whether it is on or off-centre.
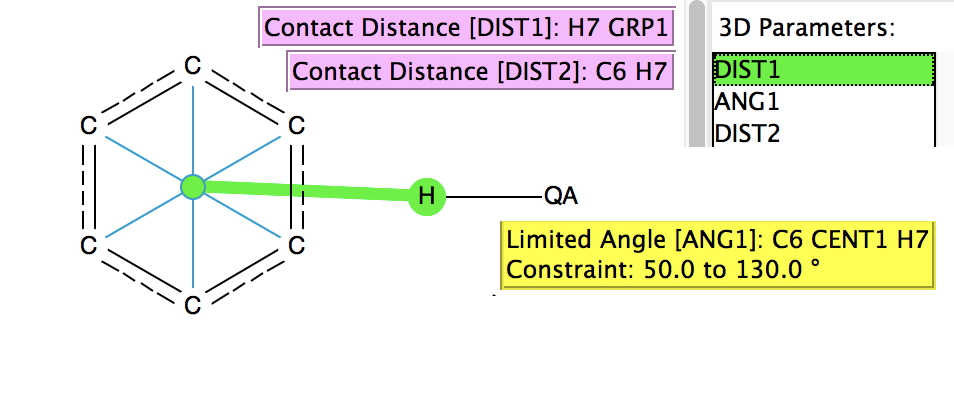
The search (February 2017 database, DOI:10.14469/hpc/2233) is shown above, QA = N, O, F, Cl and other constraints are R < 0.01, no errors, no disorder. Two distances are plotted, one (DIST1) is from the H to the ring centroid and the second (DIST2) from the H to an edge carbon atom. The colour code relates to ANG1, the angle subtended at the centroid. A value of 90° would indicate the H is orthogonal to the plane of the aromatic ring.
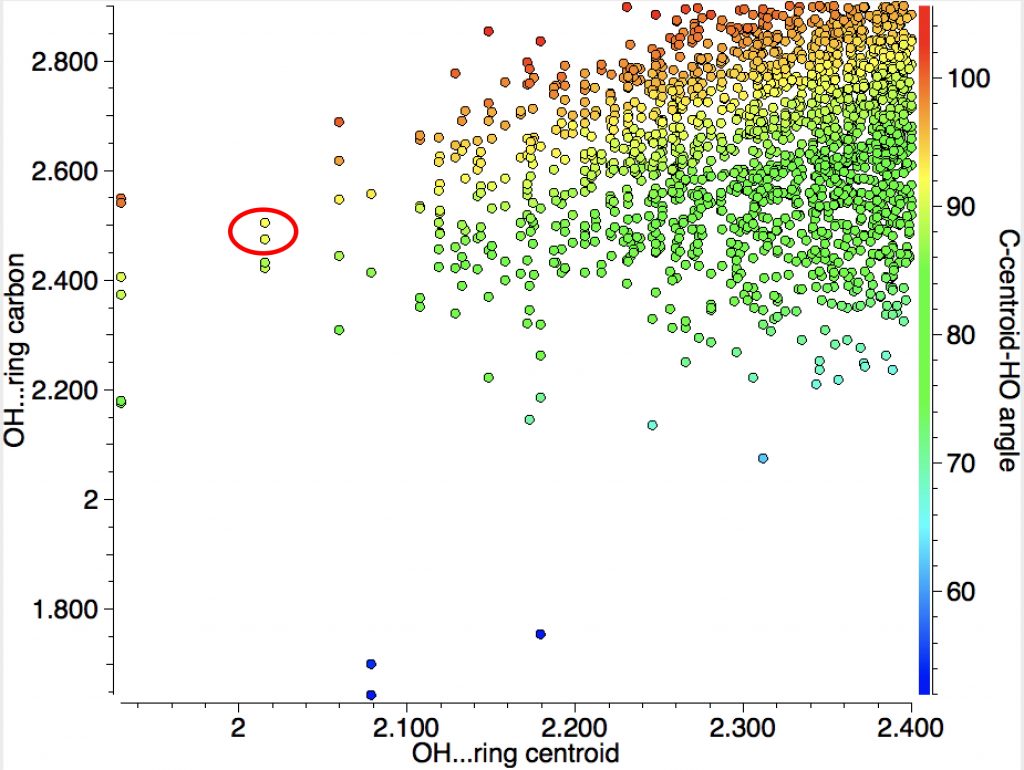
You can see from the above that the yellow dots correspond to ~90° and that by and large the H…centroid distances are shorter than the H…C distances.
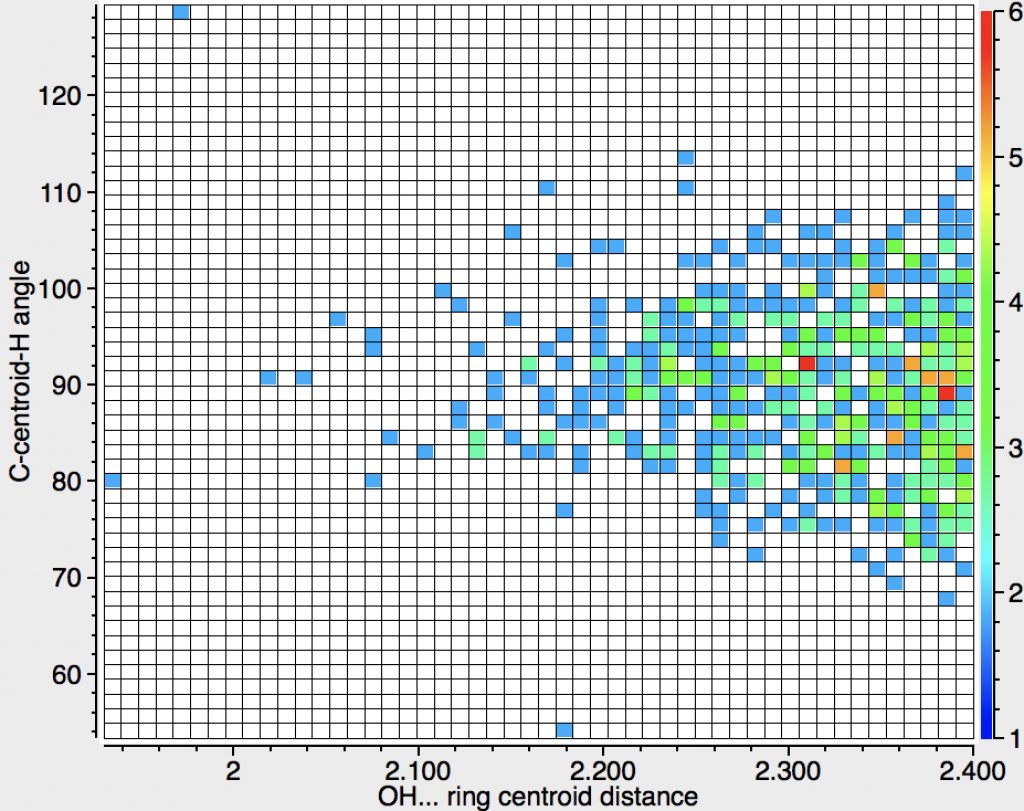
The above is another representation of this search, again showing that the preferred angle is 90°, although there is a fair bit of scatter. The extreme outliers may be crystallographic errors, but one point caught my eye and is circled in red above; ammonium tetrafluoroborate (3D model DOI: 10.5517/CC4V6TZ). This has a very short distance from the H to the centroid (2.07Å), shorter than the Pirkle reagent that we looked at all those years back. The authors[3] note that “The N-H…Ph distances, H…M 2.067Å … are exceptionally short (M = aromatic midpoint)” but also that “even at 20 K the ammonium ion performs large amplitude motions which allow the N-H vectors to sample the entire face of the aromatic system.” This implies that such bonds are largely agnostic about whether they bind to the centroid of the ring or to its edge and that the most probable position might arise simply because of crystal packing. An interesting variation on this molecule is a crystal that includes a further 5NH3 in addition to ammonium tetraphenylborate (3D model DOI: 10.5517/cc7bly2). Here an ammonia intervenes between the ammonium cation and a phenyl ring, resulting in a binding of the ammonia with two NHs closer to the edge of the ring and one NH interacting in parallel mode.
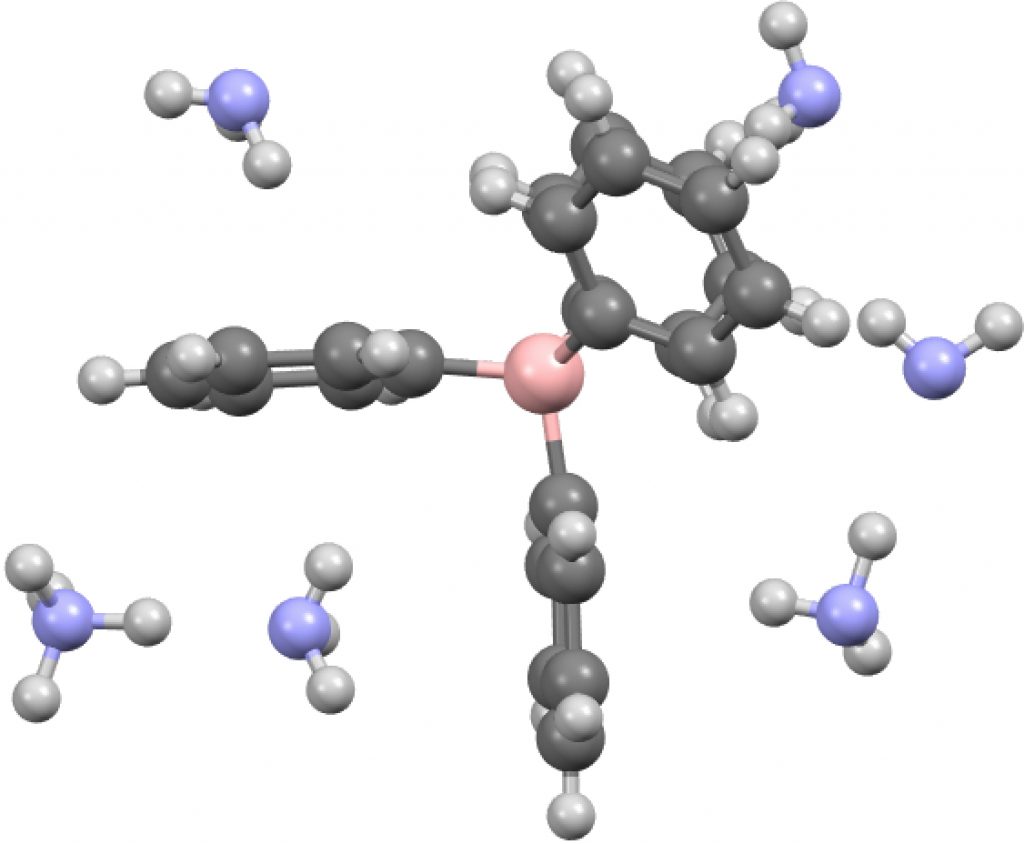
Time therefore for a calculation, using B3LYP+GD3BJ/Def2-TZVPP, the functional being chosen because the dispersion contribution is not built in, but uses what is currently thought to be the best representation of these attractions. The issue now is what molecular unit to use? This is an ionic structure and so a periodic boundary model is most appropriate, but given its size I reduced this to two models comprising smaller discrete fragments.
- A unit just comprising the simple ion pair. This leaves two of the four N-H bonds devoid of hydrogen bonding (DOI:10.14469/hpc/2234). The optimisation adopts a pose where two NH groups are directed towards a carbon atom rather than the ring centroid. How much of this is due to the smallness of this model?
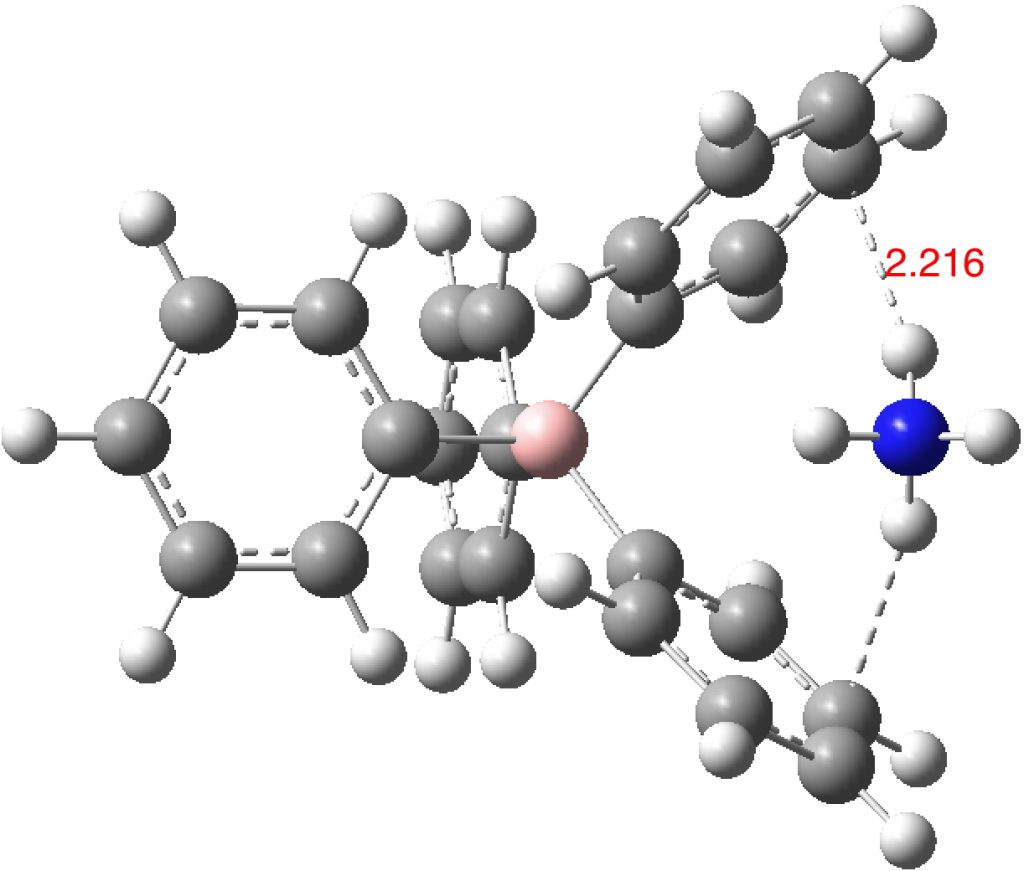
- A unit comprising a double ion pair, which allows one ammonium group to participate with all four NH groups across four phenyl rings and exhibiting six NH interactions in total with six rings (DOI: 10.14469/hpc/2235). The NH hydrogen vectors all interact with ring carbons rather than the ring centroid.
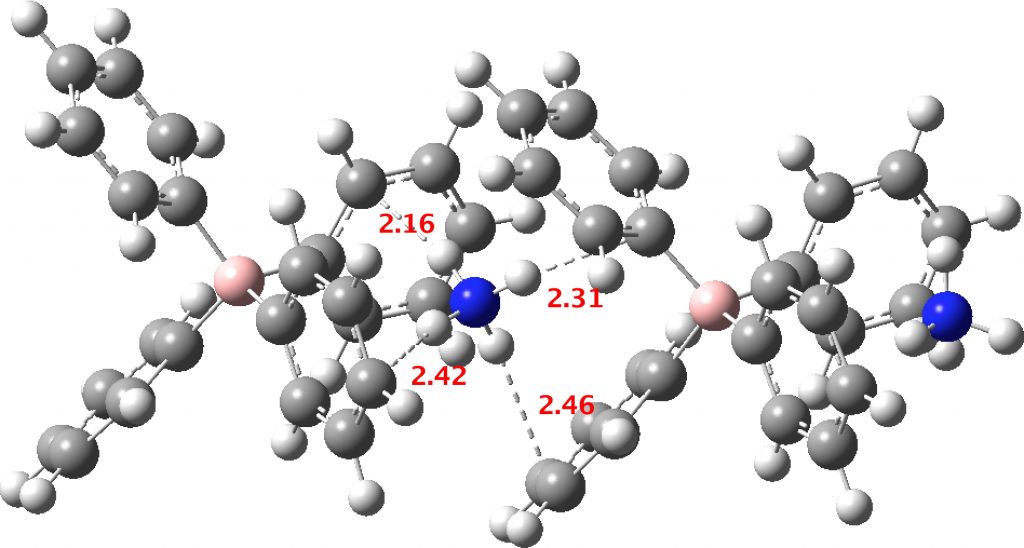
This brief computational exploration has covered only one method (the B3LYP DFT procedure), albeit with what is thought to be a good dispersion attraction term added and a reasonable basis set. It does seem to show that hydrogen bonds interacting with the centroid of a phenyl ring are not the preferred mode, which is instead an interaction with the edge of the ring. The quote above, “even at 20 K the ammonium ion performs large amplitude motions which allow the N-H vectors to sample the entire face of the aromatic system” suggests that whilst the average position might be the centroid, a true hydrogen bond to the centroid might be rarer than thought. Although most of the crystallographic examples located in the searches above deem to show a preference for the ring centroid, this might be more apparent than real.
Author
References
- H.S. Rzepa, M.L. Webb, A.M.Z. Slawin, and D.J. Williams, "? Facial hydrogen bonding in the chiral resolving agent (S)-2,2,2-trifluoro-1-(9-anthryl)ethanol and its racemic modification", Journal of the Chemical Society, Chemical Communications, pp. 765, 1991. https://doi.org/10.1039/c39910000765
- H.S. Rzepa, M.H. Smith, and M.L. Webb, "A crystallographic AM1 and PM3 SCF-MO investigation of strong OH ⋯π-alkene and alkyne hydrogen bonding interactions", J. Chem. Soc., Perkin Trans. 2, pp. 703-707, 1994. https://doi.org/10.1039/p29940000703
- T. Steiner, and S.A. Mason, "Short N<sup>+</sup>—H...Ph hydrogen bonds in ammonium tetraphenylborate characterized by neutron diffraction", Acta Crystallographica Section B Structural Science, vol. 56, pp. 254-260, 2000. https://doi.org/10.1107/s0108768199012318
Tags: Ammonia, Ammonium, aromaticity, Cations, Centroid, chemical bonding, Chemistry, Hydrogen bond, Phenyl group