The reduction of cinnamaldehyde by lithium aluminium hydride (LAH) was reported in a classic series of experiments[1],[2],[3] dating from 1947-8. The reaction was first introduced into the organic chemistry laboratories here at Imperial College decades ago, vanished for a short period, and has recently been reintroduced again.‡ The experiment is really simple in concept; add LAH to cinnamaldehyde and you get just reduction of the carbonyl group; invert the order of addition and you additionally get reduction of the double bond. Here I investigate the mechanism of these reductions using computation (ωB97XD/6-311+G(d,p)/SCRF=diethyl ether).

The mechanism can be envisaged as proceeding through a 1,4-hydride attack (TS14) with a hidden intermediate (HI14) on the reaction path, or instead finding a pathway involving either one or two consecutive 1,2-attacks; TS12-1, TS12-2 via an explicit intermediate I12. Experiment shows that quenching with D2O at the end of the reduction to replace a C-Al with a C-D bond certainly seems to rule out the 1,4 route, since that would not lead to incorporation of deuterium at the benzylic position. So does the computational model reflect this reality?
| Species | Relative ΔG, kcal/mol | FAIR Data-DOI |
|---|---|---|
| R | 0.0 | [4] |
| TS14 | +11.7 | [5] |
| P14 | -38.8 | [6] |
| TS12-1 | +8.4 | [7] |
| I12 | -35.8 | [8] |
| TS12-2 | +6.5 (42.3) | [9] |
| P12 | -52.4 | [10] |
I have chosen a model in which two dimethyl ether molecules solvate the lithium cation. The reactant itself has an interesting structure, in which two of the Al-H bonds form bridges to the Li, which ends up being five-coordinated. Further weak C-H…O=C hydrogen bonding is also observed. The NCI (non-covalent-interaction) surfaces are well worth inspecting (inspection notes: the NCI surrounding the Al has artefacts, since the value of the electron density surrounding the metal is lower than covalent density for the other elements. Click on the image below to load the 3D model).
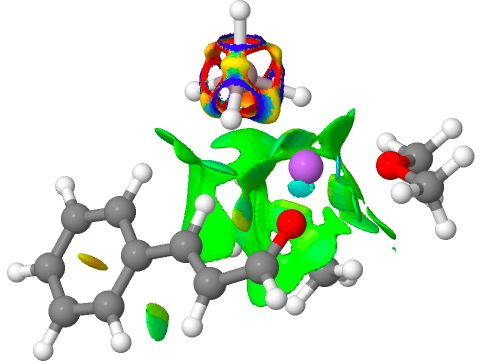
Click for 3D
TS14 retains that C-H…O=C hydrogen bond, but the double Al-H-Li bridge is lost. The 8-ring for the TS allows the hydride transfer to be approximately linear, and the Bürgi-Dunitz angle of approach of the hydride to the double bond is 107.4°. Whilst the barrier is acceptably low, the reaction reaches a cul-de-sac down this path; it has no low energy escape route.
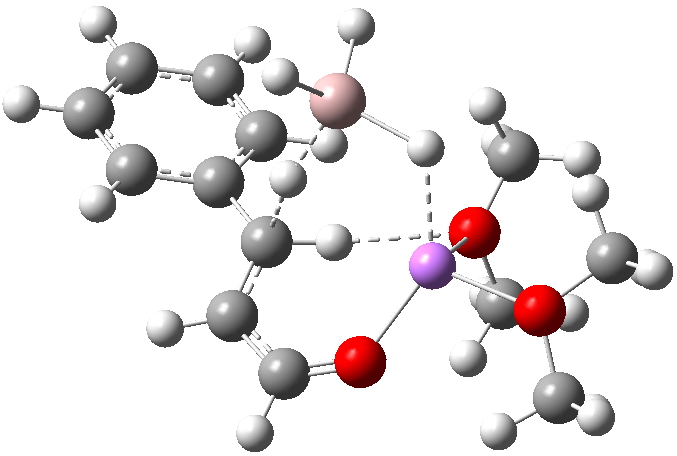
Click for 3D
TS12-1 loses the C-H…O=C hydrogen bond, but being 3.3 kcal/mol lower in free energy than TS14 fortunately provides a lower energy alternative to that cul-de-sac! The Bürgi-Dunitz angle is 112.0°.
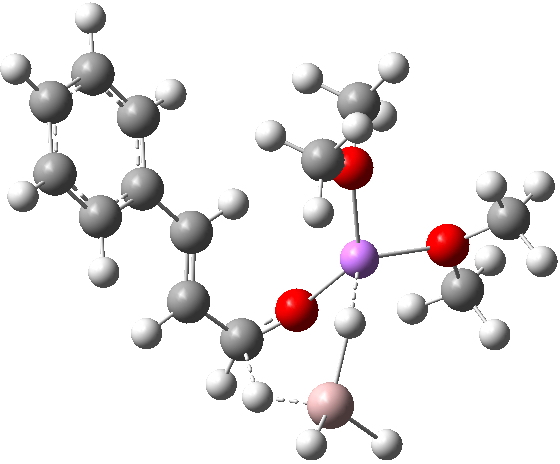
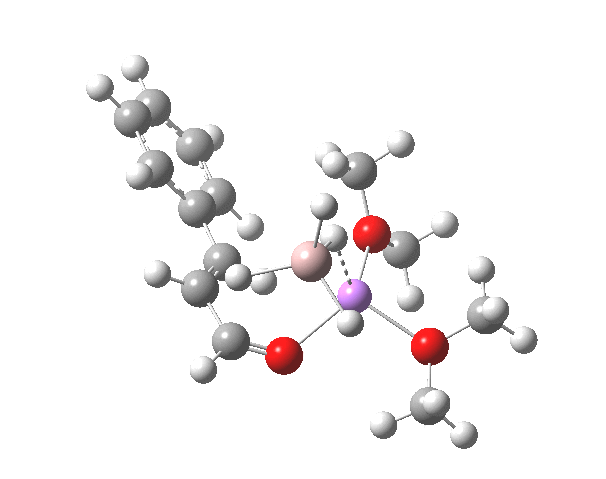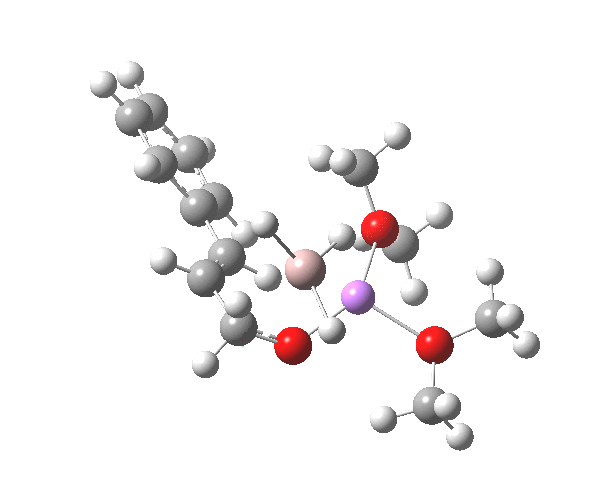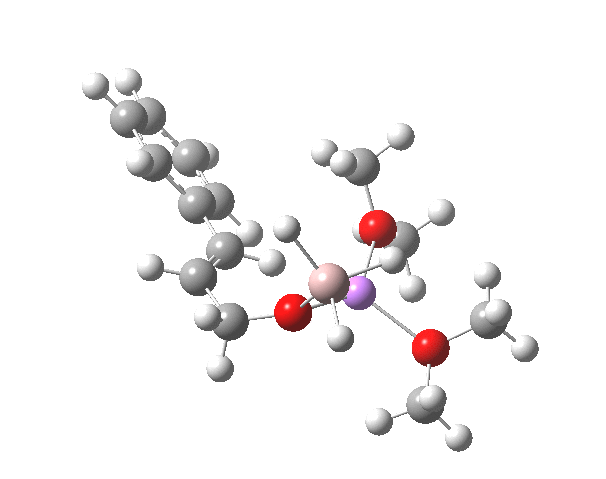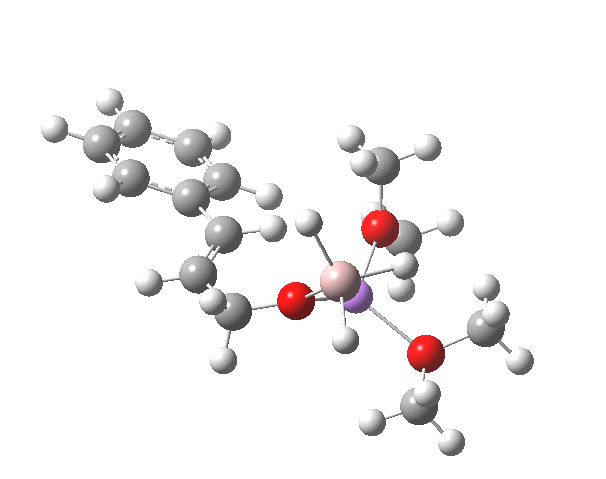
TS12-2 is required to proceed further to the dihydrocinnamyl alcohol reduction product P12, and now we have to confront the nub of the problem. Why does this further reduction only proceed when the LAH is in excess? TS12-2 itself corresponds to an Al-H addition across a C=C double bond.[11]†, with a similar barrier to TS12-1. The answer to this conundrum is to recognise that I12 forms what is called a resting state for the reaction, and that to proceed further the reaction has to overcome the barrier from I12 to TS12-2. That barrier is 42.3 kcal/mol, far too high to proceed thermally. When one encounters an unreasonable barrier, one has to look very carefully at the model one has constructed for the process.
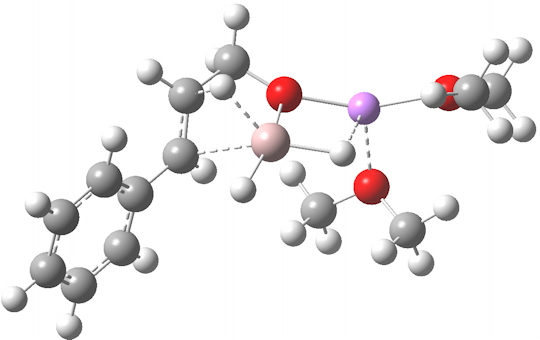
Click for 3D
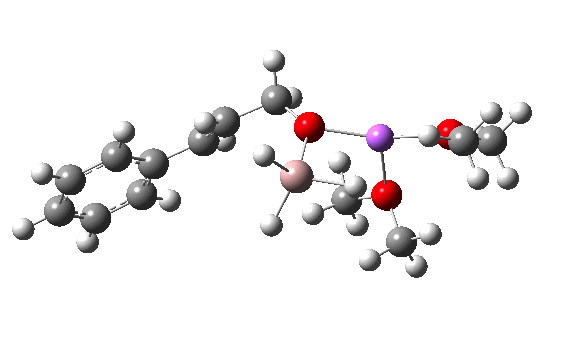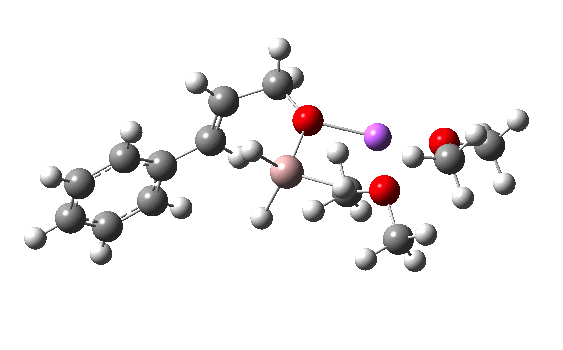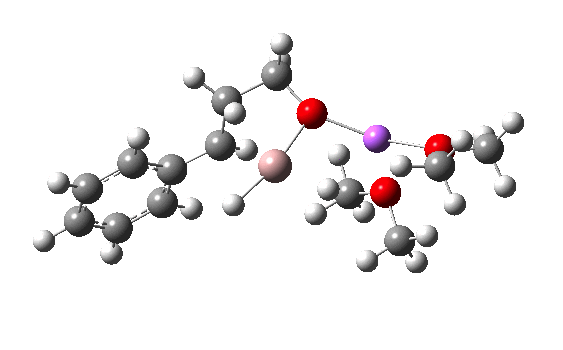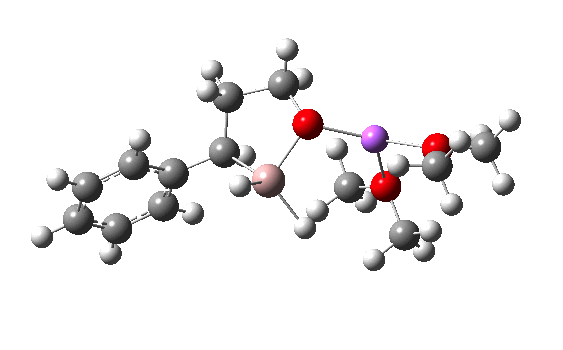
Clearly, the model I used here is lacking something. Since the reaction only proceeds when LAH is in excess, we can formulate the hypothesis that further LAH must be added to the model, from which a more reasonable barrier might emerge. If I find out how that can be done, I will report back here.
‡ LAH as a reagent was originally available in powder form, which could be quite tricky to handle and could cause fires if not handled properly. The lab organiser Chris tells me it now comes in standard-sized pellets which are far easier and safer to handle in a laboratory, allowing its re-introduction.
†Biographical note. This footnote is added because I spent three years as a Ph.D. student trying to construct transition state models by measuring kinetic isotope effects. My failure to do so convincingly meant I decided to spend a further three years as a Post Doc inverting the concept by learning how to model transition states using quantum mechanical computation. I first applied these skills as an independent researcher to locating the transition state for Cl-H addition (vs Al-H in this post) across a C=C double bond and computing the associated isotope effects.[12] This article ends with the assertion that “SCF-MO calculations may provide a more rational basis for interpreting kinetic isotopes than the reverse procedure of attempting to establish a transition state model from the observed kinetic data.” It is nice to see that posterity has shown that this assessment was about right.
Author
References
- R.F. Nystrom, and W.G. Brown, "Reduction of Organic Compounds by Lithium Aluminum Hydride. I. Aldehydes, Ketones, Esters, Acid Chlorides and Acid Anhydrides", Journal of the American Chemical Society, vol. 69, pp. 1197-1199, 1947. https://doi.org/10.1021/ja01197a060
- R.F. Nystrom, and W.G. Brown, "Reduction of Organic Compounds by Lithium Aluminum Hydride. II. Carboxylic Acids", Journal of the American Chemical Society, vol. 69, pp. 2548-2549, 1947. https://doi.org/10.1021/ja01202a082
- F.A. Hochstein, and W.G. Brown, "Addition of Lithium Aluminum Hydride to Double Bonds", Journal of the American Chemical Society, vol. 70, pp. 3484-3486, 1948. https://doi.org/10.1021/ja01190a082
- H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191154
- H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191148
- H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191152
- H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191149
- H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191151
- H.S. Rzepa, and H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191156
- H.S. Rzepa, "C 13 H 24 Al 1 Li 1 O 3", 2015. https://doi.org/10.14469/ch/191155
- H.S. Rzepa, "Gaussian Job Archive for C2H7Al", 2015. https://doi.org/10.6084/m9.figshare.1362146
- H.S. Rzepa, "MNDO SCF-MO calculations of kinetic isotope effects for dehydrochlorination reactions of chloroalkanes", Journal of the Chemical Society, Chemical Communications, pp. 939, 1981. https://doi.org/10.1039/c39810000939
Tags: Al-H-Li bridge, dihydrocinnamyl alcohol reduction product, free energy, Imperial College, independent researcher, low energy escape route, lower energy alternative, metal, pence
I would be very interested to learn about the mechanism. A clue might come from the related hydroalumination of propargyl alcohols, which is a useful method to prepare trisubstituted alkenes, stereoselectively.
The early key paper is E. J. Corey et al, JACS 1967: DOI: 10.1021/ja00992a065 (http://pubs.acs.org/doi/pdf/10.1021/ja00992a065)
In that case, the hydroalumination proceeds by anti-addition of aluminum and hydrogen across the triple bond to give (E)-alkenes, after protic quench. This information about the stereochemistry of addition is not readily available in case of the allylic alcohol, where your first mechanism (with the overly high energy barrier) above assumes syn-addition.
The hint from the propargyl alcohol hydroalumination could be that maybe the addition is anti for cinnamyl alcohol, too? Of course, that could imply why an excess of reagent is needed…
There is another detail in the propargyl alcohol hydroalumination case: depending on the reagent used (a: LiAlH4 + AlCl3; b: LiAlH4 + NaOMe), the aluminum-species formed has the metal at the beta- (a) or the gamma- (b) position (relative to the propargyl-C-OH = alpha), but in either case the addition proceeds anti… (see the Corey paper). This would imply that either a four- (a) or five- (b) membered alumina-oxa-cycle is formed by anti-addition? Or maybe not a cyclic mechanism, after all.
Not sure if this has been investigated mechanistically. It is one of those reactions often used, but little understood. (Actually, I now see that the book by Clayden et al (Organic Chemistry, 2012 edition) proposes a mechanism on page 683 with one aluminum as Lewis acid, the other as hydride donor – it might be speculation?).
Maybe you can enlighten us?
Yes, anti-addition of hydride by a second AlH4 to I12, with concomitant (concerted or stepwise) benzylic C-Al bond formation is the most obvious transition state to go hunting. This will involve adding a second AlH4(-).Li(OR)2 (or AlH4(-).Li(OR)3 ?) to the system, which starts to slow the computation down significantly.
When (if) I get a result, I will report back!
I would make the more general point that textbooks such as eg Clayden, tend to produce mechanisms based on reasonable arrow pushing (and perhaps kinetics), but to my knowledge no student text has yet incorporated quantum computation to accompany these speculations. I would make a further prediction that such a text WILL emerge in the next decade (and no, I am not writing one now!!).
A good approach when seeking inspiration for more complex systems is to raid the crystal structure databases. Here are structures found there which contain two Al.Li units, with various solvents as ligands. The structural diversity is large!
I remember doing this very reaction as an undergrad at IC in 1978!
A rather dumb question, but I’ll ask it anyway: has anyone tried treating the allylic alcohol with LAH to see if it is hydroaluminated? Perhaps some careful kinetic work could determine whether one or two LAH’s are needed for the second step to go.
Nick,
If you read Ref 3 in the blog, you will find they report adding LAH to cinnamyl alcohol initially reacts with the OH group in a very fast reaction, and then further addition of LAH more slowly eliminates H2 gas, with hydrocinnamyl alcohol recovered after “hydrolytic decomposition”. They speculate about the nature of the aluminate, but it is not characterised. The crystal database contains nothing resembling the product P12 above.