Another foray into one of the more famous anecdotal chemistry “models”, the analysis of which led directly to the formulation of the WoodWard-Hoffmann (stereochemical) rules for pericyclic reactions. Previously, I tried to produce a modern computer model of what Woodward might have had to hand when discovering that the stereochemical outcome of a key reaction in his vitamin B12 synthesis was opposite to that predicted using his best model of the reaction.
Archive for the ‘pericyclic’ Category
The thermal reactions … took precisely the opposite stereochemical course to that which we had predicted. A non-covalent-interaction view of the model.
Wednesday, February 3rd, 2021The thermal reactions … took precisely the opposite stereochemical course to that which we had predicted
Wednesday, January 20th, 2021The quote of the post title comes from R. B. Woodward explaining the genesis of the discovery of what are now known as the Woodward-Hoffmann rules for pericyclic reactions.[1] I first wrote about this in 2012, noting that “for (that) blog, I do not want to investigate the transition states”. Here I take a closer look at this aspect.
References
- R.B. Woodward, and R. Hoffmann, "Stereochemistry of Electrocyclic Reactions", Journal of the American Chemical Society, vol. 87, pp. 395-397, 1965. https://doi.org/10.1021/ja01080a054
Trimerous pericyclic reactions: what is the effect of changing the electron count by two?
Monday, November 2nd, 2020In an earlier post, I pondered on how the “arrow pushing” for the thermal pericyclic reactions of some annulenes (cyclic conjugated hydrocarbons) could be represented in terms of either two separate electrocyclic reactions or of one cycloaddition reaction. Each reaction is governed by selection rules which can be stated in terms of the anticipated aromaticity of the pericyclic transition state as belonging to a 4n or a 4n+2 class. This in turn determines whether the topology of the transition state belongs to a class of aromatic species known as either Hückel or Möbius. Here I play with the observation that by adding or removing two electrons from the molecule, the two classes 4n and 4n+2 can be swapped. What happens to the aromaticities of the transition states if that is done?
Trimerous pericyclic reactions.
Thursday, October 8th, 2020I occasionally spot an old blog that emerges, if only briefly, as “trending”. In this instance, only the second blog I ever wrote here, way back in 2009 as a follow up to this article.[1] With something of that age, its always worth revisiting to see if any aspect needs updating or expanding, given the uptick in interest. It related to the observation that there can be more than one way of expressing the “curly arrows” for some pericyclic reactions. These alternatives may each represent different types of such reactions, hence leading to a conundrum for students of how to label the mechanism. I had noted in that blog that I intended to revisit the topic and so a mere eleven years later here it is!
References
- H.S. Rzepa, "The Aromaticity of Pericyclic Reaction Transition States", Journal of Chemical Education, vol. 84, pp. 1535, 2007. https://doi.org/10.1021/ed084p1535
Dyotropic Ring Expansion: more mechanistic reality checks.
Sunday, October 1st, 2017I noted in my WATOC conference report a presentation describing the use of calculated reaction barriers (and derived rate constants) as mechanistic reality checks. Computations, it was claimed, have now reached a level of accuracy whereby a barrier calculated as being 6 kcal/mol too high can start ringing mechanistic alarm bells. So when I came across this article[1] in which calculated barriers for a dyotropic ring expansion observed under mild conditions in dichloromethane as solvent were used to make mechanistic inferences, I decided to explore the mechanism a bit further.
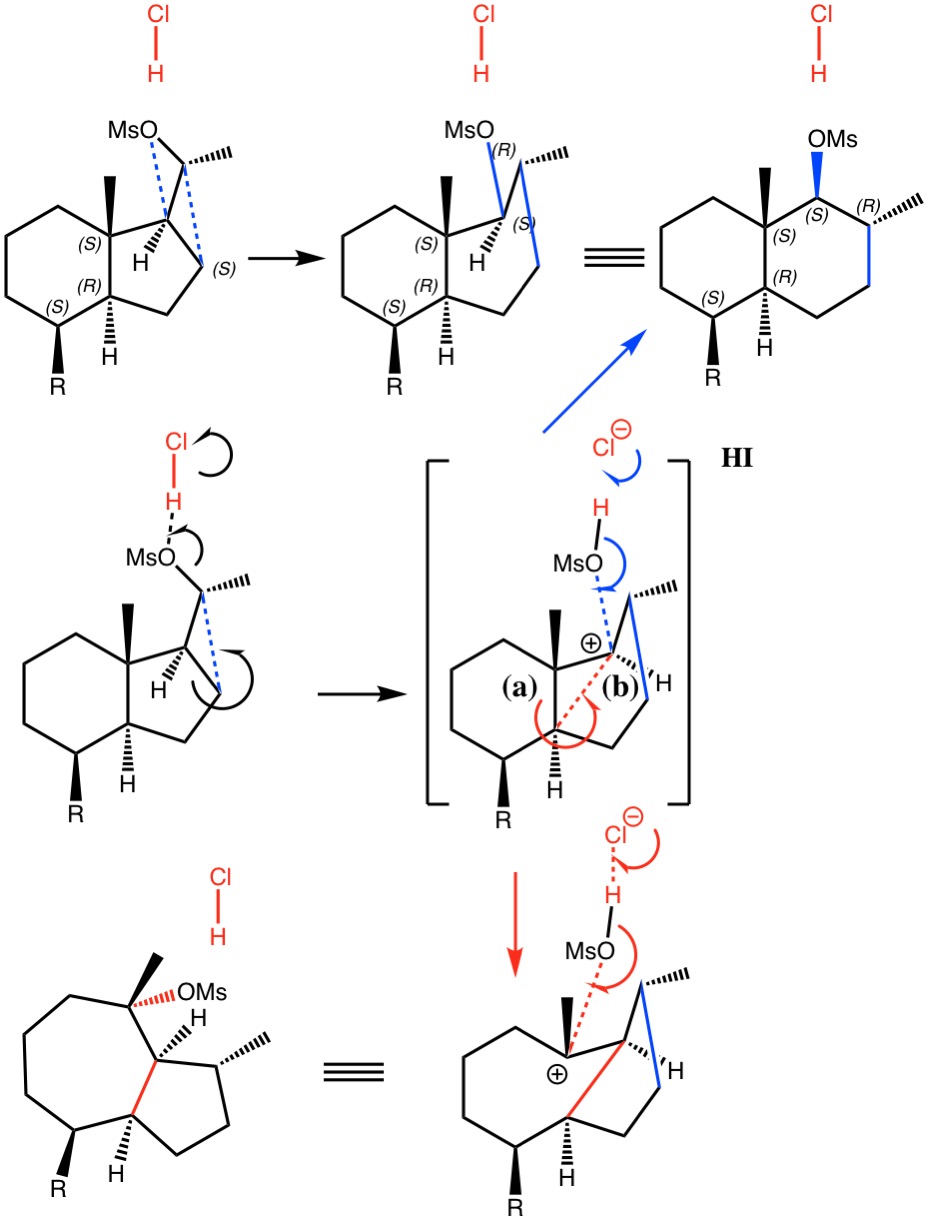
References
- H. Santalla, O.N. Faza, G. Gómez, Y. Fall, and C. Silva López, "From Hydrindane to Decalin: A Mild Transformation through a Dyotropic Ring Expansion", Organic Letters, vol. 19, pp. 3648-3651, 2017. https://doi.org/10.1021/acs.orglett.7b01621
Computationally directed synthesis: 2,3-dimethyl-2-butene + NO(+).
Saturday, September 6th, 2014In the previous posts, I explored reactions which can be flipped between two potential (stereochemical) outcomes. This triggered a memory from Alex, who pointed out this article from 1999[1] in which the nitrosonium cation as an electrophile can have two outcomes A or B when interacting with the electron-rich 2,3-dimethyl-2-butene.  NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1]
NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1] 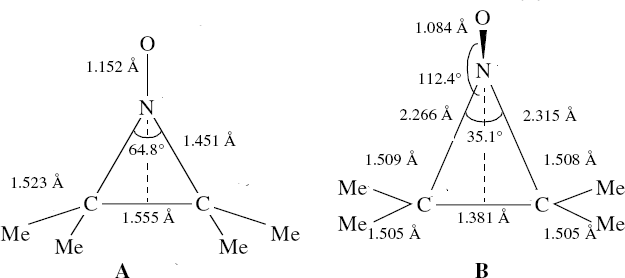 Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest.
Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest. 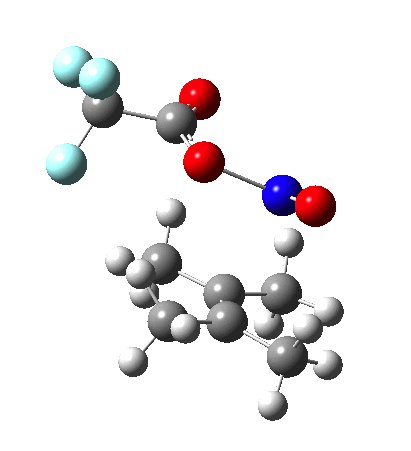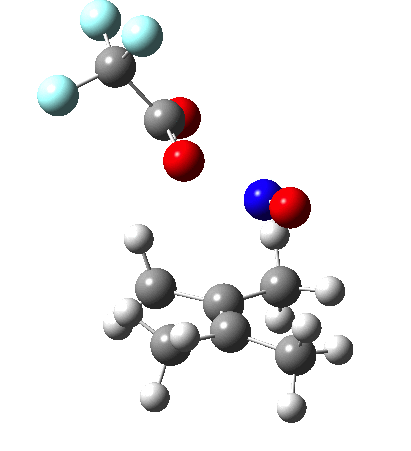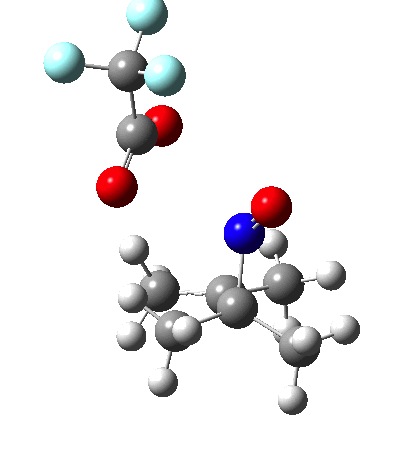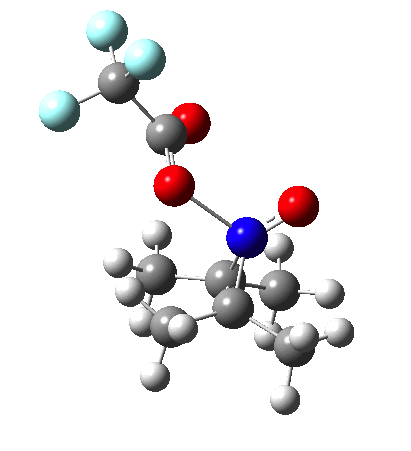

 Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.
Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.  I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate).
I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate). 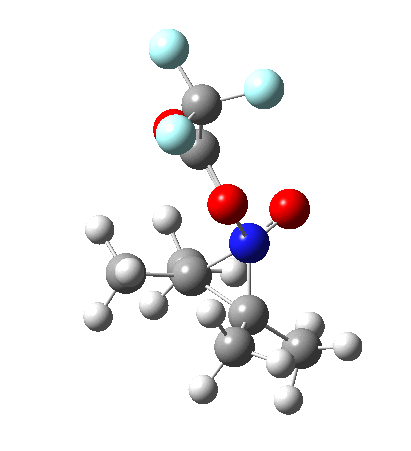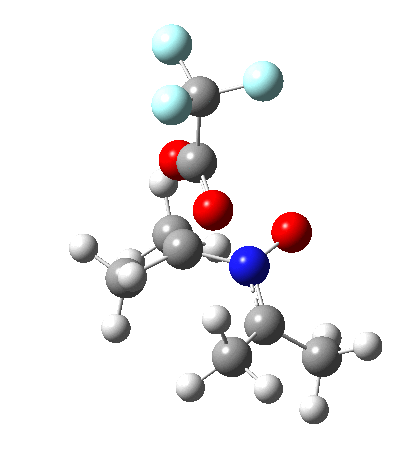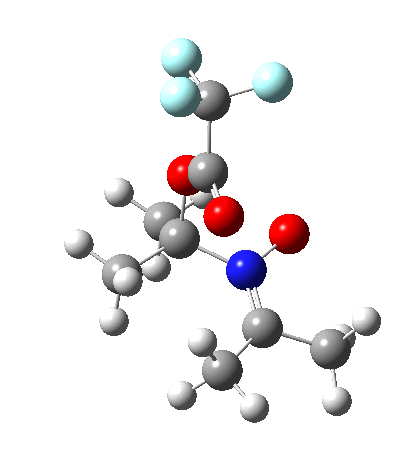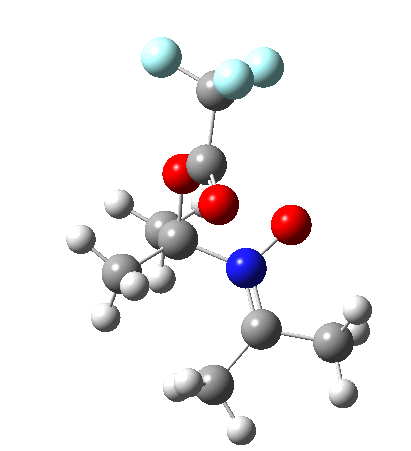
 So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
References
- G.I. Borodkin, I.R. Elanov, A.M. Genaev, M.M. Shakirov, and V.G. Shubin, "Interaction in olefin–NO+ complexes: structure and dynamics of the NO+–2,3-dimethyl-2-butene complex", Mendeleev Communications, vol. 9, pp. 83-84, 1999. https://doi.org/10.1070/mc1999v009n02abeh000995
- H.S. Rzepa, "C8H12F3NO3", 2014. https://doi.org/10.14469/ch/24979
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162797
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162676
Full circle. Stereoisomeric transition states for [1,4] pericyclic shifts.
Monday, August 18th, 2014This post, the fifth in the series, comes full circle. I started off by speculating how to invert the stereochemical outcome of an electrocyclic reaction by inverting a bond polarity. This led to finding transition states for BOTH outcomes with suitable substitution, and then seeking other examples. Migration in homotropylium cation was one such, with the “allowed/retention” transition state proving a (little) lower in activation energy than the “forbidden/inversion” path. Here, I show that with two electrons less, the stereochemical route indeed inverts. First, a [1,4] alkyl shift with inversion at the migrating carbon (ωB97XD/6-311G(d,p)/SCRF=chloroform); as a four-electron process, this is the “allowed” route.[1]
First, a [1,4] alkyl shift with inversion at the migrating carbon (ωB97XD/6-311G(d,p)/SCRF=chloroform); as a four-electron process, this is the “allowed” route.[1] 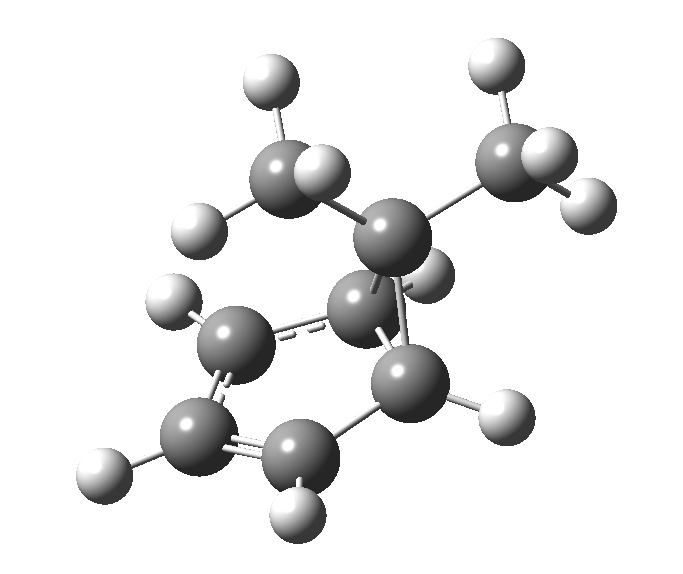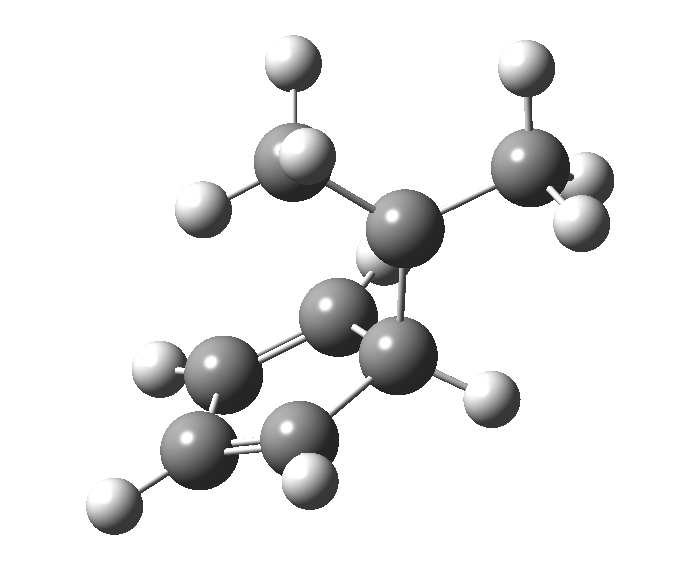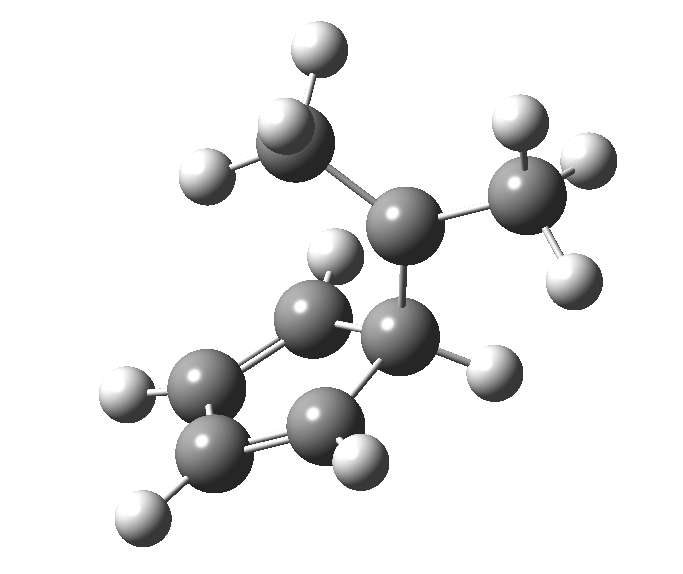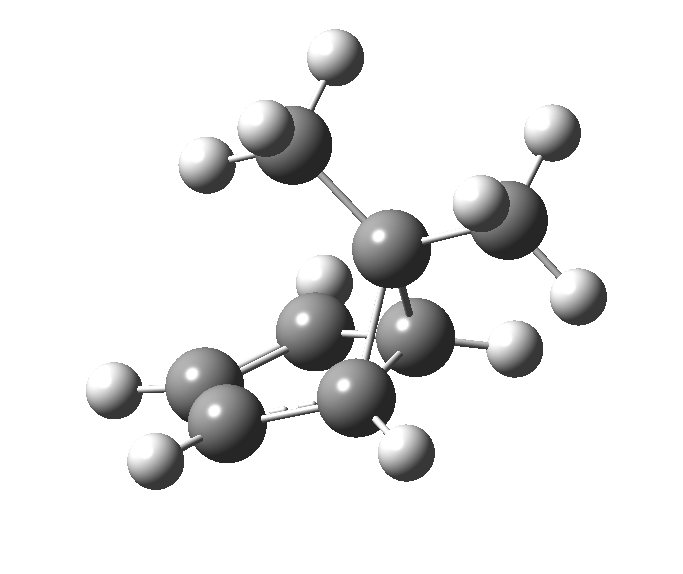 The “forbidden” route corresponds to retention of configuration at the migrating carbon.[2]
The “forbidden” route corresponds to retention of configuration at the migrating carbon.[2] 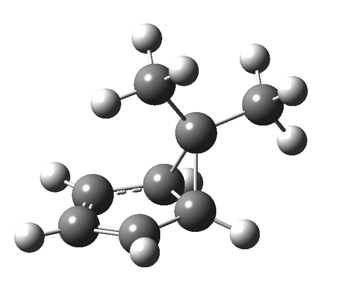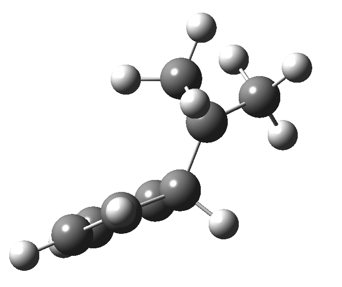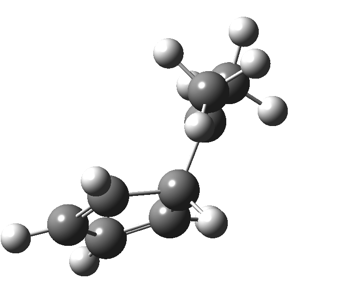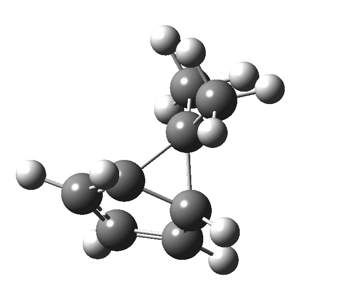 The barriers for each process can be seen below from the IRCs. That for inversion is ~4.5 kcal/mol lower than retention. This nicely transposes the values for the six-electron homologue shown in the previous post.
The barriers for each process can be seen below from the IRCs. That for inversion is ~4.5 kcal/mol lower than retention. This nicely transposes the values for the six-electron homologue shown in the previous post. 
 There is one more nugget of insight that can be extracted. The start/end-point for the six-electron process (homotropylium cation) was, as the name implies, homoaromatic. Now, with a four-electron system we also have an inverse. Nominally, we should now end with homo-antiaromaticity (but see [3]). But antiaromaticity is avoided whenever possible, and so the homoaromatic bond observed in homotropylium is not formed. It resolutely remains a σ-bond (1.48Å) thus sequestering two electrons, and the remaining two electrons simply form a delocalised allyl cation. With the six-electron homotropylium, reactant/product were stabilised by that additional (homo)aromaticity, thus inducing a relatively high barrier. With the four-electron system here, no such reactant/product stabilisation occurs, and hence the reaction barriers are now significantly lower. A rather neat pedagogic example.
There is one more nugget of insight that can be extracted. The start/end-point for the six-electron process (homotropylium cation) was, as the name implies, homoaromatic. Now, with a four-electron system we also have an inverse. Nominally, we should now end with homo-antiaromaticity (but see [3]). But antiaromaticity is avoided whenever possible, and so the homoaromatic bond observed in homotropylium is not formed. It resolutely remains a σ-bond (1.48Å) thus sequestering two electrons, and the remaining two electrons simply form a delocalised allyl cation. With the six-electron homotropylium, reactant/product were stabilised by that additional (homo)aromaticity, thus inducing a relatively high barrier. With the four-electron system here, no such reactant/product stabilisation occurs, and hence the reaction barriers are now significantly lower. A rather neat pedagogic example.
References
- H.S. Rzepa, "Gaussian Job Archive for C8H11(1+)", 2014. https://doi.org/10.6084/m9.figshare.1142175
- H.S. Rzepa, "Gaussian Job Archive for C8H11(1+)", 2014. https://doi.org/10.6084/m9.figshare.1142174
- C.S.M. Allan, and H.S. Rzepa, "Chiral Aromaticities. A Topological Exploration of Möbius Homoaromaticity", Journal of Chemical Theory and Computation, vol. 4, pp. 1841-1848, 2008. https://doi.org/10.1021/ct8001915
An unusual [1,6] shift in homotropylium cation exhibiting zones of aromaticity.
Tuesday, August 12th, 2014One thing leads to another. Thus in the previous post, I described a thermal pericyclic reaction that appears to exhibit two transition states resulting in two different stereochemical outcomes. I noted that another such reaction appeared to be a [1,6] carousel migration in homotropylium cation,[1] where transition states for both retention and inversion of the configuration of the migrating group (respectively formally allowed and forbidden) were reported (scheme below). Here I explore this system further.  Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array.
Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array. 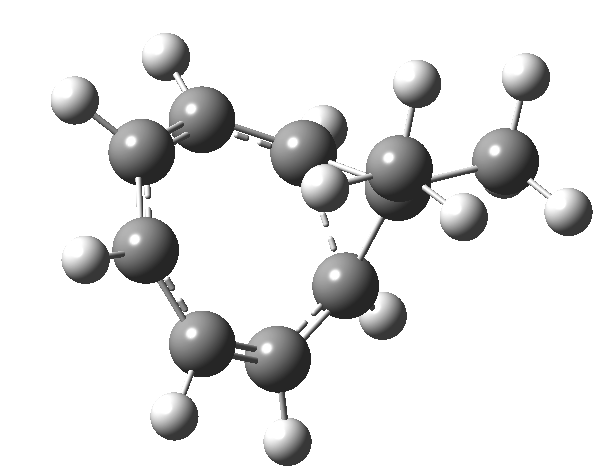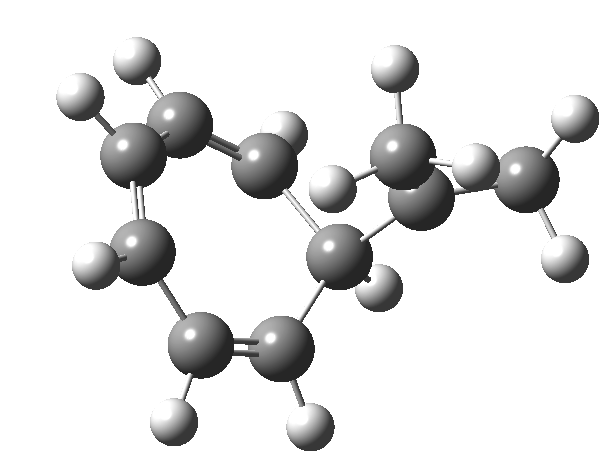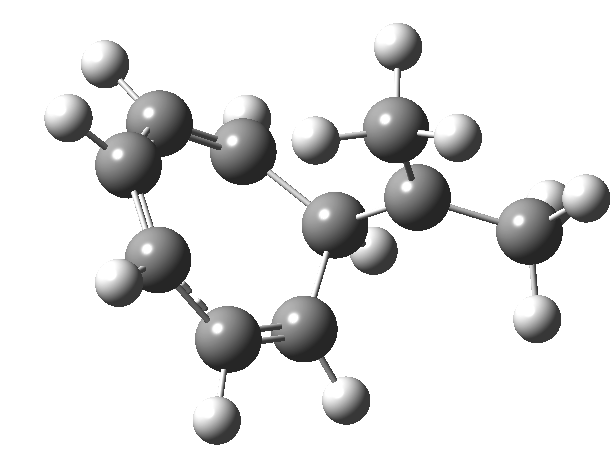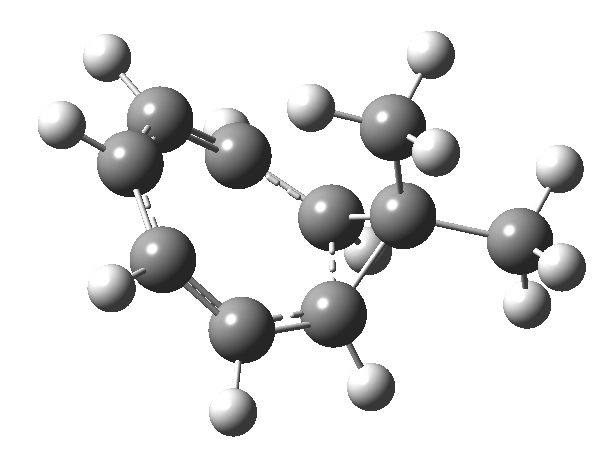

 The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
References
- A.M. Genaev, G.E. Sal’nikov, and V.G. Shubin, "Energy barriers to carousel rearrangements of carbocations: Quantum-chemical calculations vs. experiment", Russian Journal of Organic Chemistry, vol. 43, pp. 1134-1138, 2007. https://doi.org/10.1134/s1070428007080076
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1134556
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135694
Using a polar bond to flip: on the knife-edge!
Sunday, August 10th, 2014In my first post on the topic, I discussed how inverting the polarity of the C-X bond from X=O to X=Be (scheme below) could flip the stereochemical course of the electrocyclic pericyclic reaction of a divinyl system. This was followed up by exploring what happens at the half way stage, i.e. X=CH2, the answer being that one gets an antarafacial pathway as with X=O. Here I fill in another gap, X=BH to see if a metaphorical microscope can be used to view the actual region of the “flip” to a suprafacial mode. This time, uniquely, it proved possible to locate TWO transition states for this process, one suprafacial[1] and one antarafacial[2], this latter being 10.5 kcal/mol lower in ΔG† (ωB97XD/6-311G(d,p)/SCRF=dichloromethane). It is quite rare to be able to find BOTH stereochemical outcomes of a thermal pericyclic reaction.‡
This time, uniquely, it proved possible to locate TWO transition states for this process, one suprafacial[1] and one antarafacial[2], this latter being 10.5 kcal/mol lower in ΔG† (ωB97XD/6-311G(d,p)/SCRF=dichloromethane). It is quite rare to be able to find BOTH stereochemical outcomes of a thermal pericyclic reaction.‡
References
- H.S. Rzepa, "Gaussian Job Archive for C5H7B", 2014. https://doi.org/10.6084/m9.figshare.1133933
- H.S. Rzepa, "Gaussian Job Archive for C5H7B", 2014. https://doi.org/10.6084/m9.figshare.1133934
Using a polar bond to flip: a follow up project.
Wednesday, August 6th, 2014In my earlier post on the topic, I discussed how inverting the polarity of the C-X bond from X=O to X=Be could flip the stereochemical course of the electrocyclic pericyclic reaction of a divinyl system. An obvious question would be: what happens at the half way stage, ie X=CH2? Well, here is the answer. 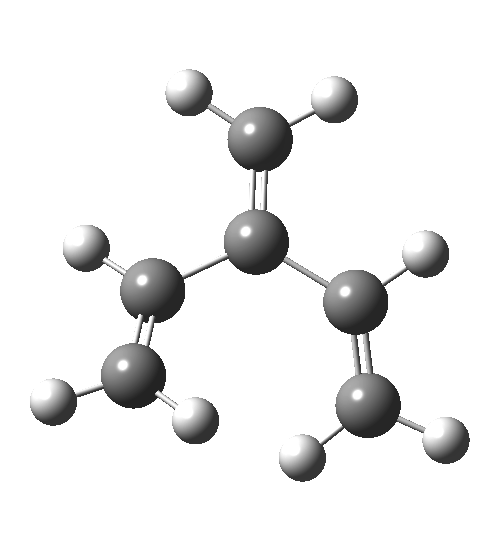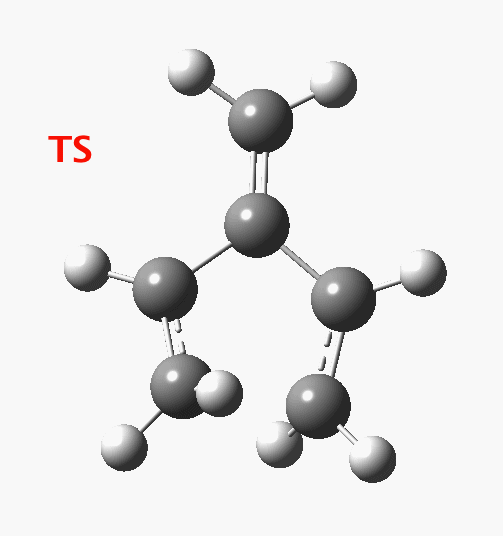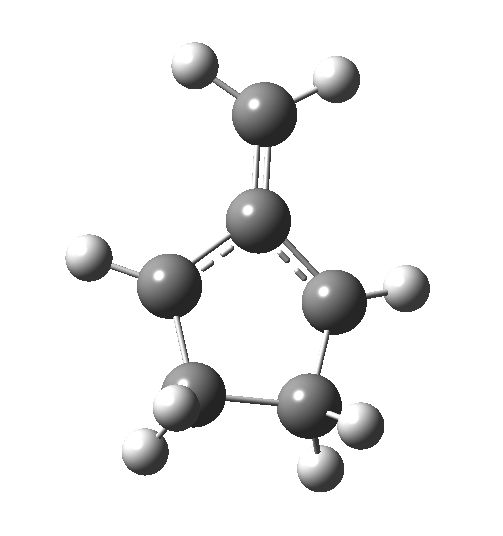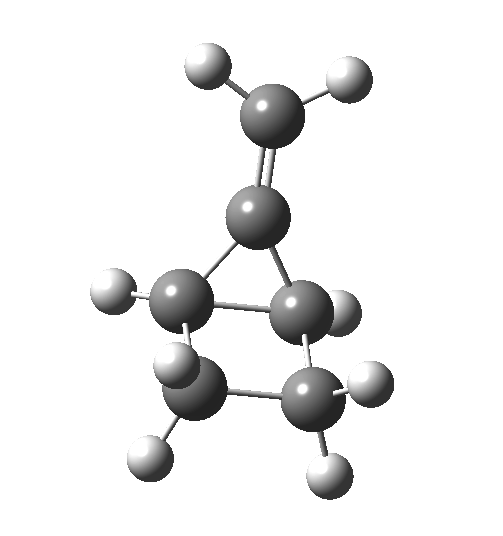 The reaction occurs in two stages (ωB97XD/6-311G(d,p)/SCRF=dichloromethane)[1] but overall is a concerted, albeit asynchronous, reaction. The initial stage is a conrotatory ring closure (as observed with X=O but opposite to X=Be), and reaching what we will call a HI (hidden intermediate). This HI clearly has zwitterionic character, and manifests its presence most obviously at IRC = -3.5 below.
The reaction occurs in two stages (ωB97XD/6-311G(d,p)/SCRF=dichloromethane)[1] but overall is a concerted, albeit asynchronous, reaction. The initial stage is a conrotatory ring closure (as observed with X=O but opposite to X=Be), and reaching what we will call a HI (hidden intermediate). This HI clearly has zwitterionic character, and manifests its presence most obviously at IRC = -3.5 below. 
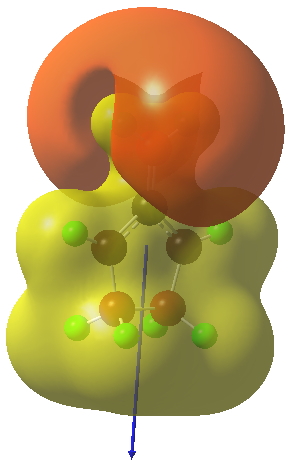
 The polarity of this HI is revealed by the dipole moment (6D) and molecular electrostatic potentials, below. The dipole vector goes from -ve to +ve, and the MEP clearly reveals the polarity below.
The polarity of this HI is revealed by the dipole moment (6D) and molecular electrostatic potentials, below. The dipole vector goes from -ve to +ve, and the MEP clearly reveals the polarity below. 
 This ionic HI however is not stable, and in the second stage of the reaction collapses to the neutral bicyclic hydrocarbon shown below. Overall, it amounts to a 2+2 cycloaddition, but with a very unusual pathway in which one C-C bond is very much formed before the other (which is how the reaction escapes the clutches of the Woodward-Hoffmann forbidden-ness).
This ionic HI however is not stable, and in the second stage of the reaction collapses to the neutral bicyclic hydrocarbon shown below. Overall, it amounts to a 2+2 cycloaddition, but with a very unusual pathway in which one C-C bond is very much formed before the other (which is how the reaction escapes the clutches of the Woodward-Hoffmann forbidden-ness).  Why is all this worth this follow-up? Well, one can now start to “design” the reaction. All three carbon atoms with formal charges can be stabilised or destabilised with appropriate substituents. It should not be too difficult to stabilise out the HI into just an I(intermediate), or indeed to remove it from the profile. Nice perhaps for a group of students, who can partition up the substituents amongst themselves and discover if they have the desired effect. And would any of this tinkering change the stereochemical outcome?
Why is all this worth this follow-up? Well, one can now start to “design” the reaction. All three carbon atoms with formal charges can be stabilised or destabilised with appropriate substituents. It should not be too difficult to stabilise out the HI into just an I(intermediate), or indeed to remove it from the profile. Nice perhaps for a group of students, who can partition up the substituents amongst themselves and discover if they have the desired effect. And would any of this tinkering change the stereochemical outcome?
References
- H.S. Rzepa, "Gaussian Job Archive for C6H8", 2014. https://doi.org/10.6084/m9.figshare.1128205