I have written earlier about dihydrocostunolide, and how in 1963 Corey missed spotting the electronic origins of a key step in its synthesis.[1]. A nice juxtaposition to this failed opportunity relates to Woodward’s project at around the same time to synthesize vitamin B12. The step in the synthesis that caused him to ponder is shown below.

In the 1950s, Linus Pauling was the shining example in the use of model building in chemistry, and the so-called CPK (Corey, Pauling and Koltun) model was being adopted by most synthetic chemists as a part of the design of their syntheses (I have argued that the progenitor of the CPK model was in fact created by Loschmidt, in 1860). These were physical models, and it is quite likely that Woodward would have used one to ponder the conversion shown above as G ⇒ J or H. As you can read from the quote above (taken from Chem. Soc. Special Publications (Aromaticity), 1967, 21, 217, a document not available online), he had concluded that G ⇒ J was more likely than G ⇒ H, and so was considerably surprised when the reaction actually proceeded to give the latter and not the former. In fact, photolysis of (the undesired) H gave I, which then did give (the desired) J upon heating, so he got what he wanted in the end (he usually did!). Of course, we now know that this electrocyclisation proceeds under what is sometimes called orbital control (as explained by Woodward and Hoffmann[2]) and what can also be taught as a manifestation of transition state aromaticity[3].
For this blog, I do not want to investigate the transition states, but just to update Woodward’s use of physical (possibly CPK) models to predict the outcome of reactions. CPK models are characterised by their use of van der Waals radii for the atom spheres, the so-called space-filling representation, and as such they are in effect looking at the repulsive steric interactions (the 12 of the 6-12 potential). What they do not do is measure the attractive dispersion contributions to the model. I had suggested that differential dispersion contributions may be a (dominant?) factor in explaining why Sharpless epoxidation goes enantioselectively. With this in mind, I optimized the geometry of species H and J above at a dispersion and solvent-corrected level of theory (ωB97XD/6-311G(d,p)/SCRF=dichloromethane) to see if the relative stabilities of the products might agree with Woodward’s prediction that J should have formed.
| ΔG 0.0 kcal/mol | ΔG +1.0 kcal/mol |
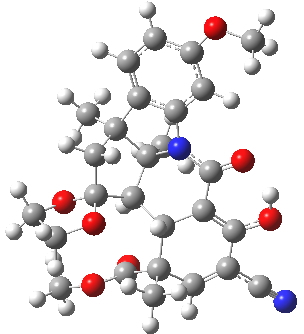 H. Click for 3D |
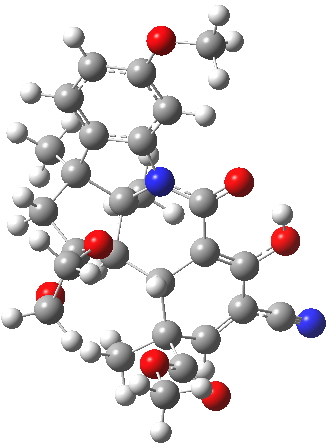 J. Click for 3D. |
This computation shows that H is the lower in free energy by 1.0 kcal/mol, and by 0.8 kcal/mol in dispersion energy. So Woodward’s hypothesis that J was the more likely to form on steric grounds is not supported by this modern equivalent of a CPK model. I should add that a CPK model may only take an hour or so to build (but possibly weeks to order the components) whereas this quantum model took around 9 hours to compute.
Author
References
- E.J. Corey, and A.G. Hortmann, "The Total Synthesis of Dihydrocostunolide", Journal of the American Chemical Society, vol. 87, pp. 5736-5742, 1965. https://doi.org/10.1021/ja00952a037
- R.B. Woodward, and R. Hoffmann, "Stereochemistry of Electrocyclic Reactions", Journal of the American Chemical Society, vol. 87, pp. 395-397, 1965. https://doi.org/10.1021/ja01080a054
- H.S. Rzepa, "The Aromaticity of Pericyclic Reaction Transition States", Journal of Chemical Education, vol. 84, pp. 1535, 2007. https://doi.org/10.1021/ed084p1535
Tags: dispersion energy, free energy, Historical, pericyclic