If you search e.g. Scifinder for N,O-diphenyl hydroxylamine (RN 24928-98-1) there is just one literature citation, to a 1962 patent. Nothing else; not even a calculation (an increasing proportion of the molecules reported in Chemical Abstracts have now only ever been subjected to calculation, not synthesis). A search of Reaxys also offers only one hit[1] reporting one unsuccessful attempt in 1963 to prepare this compound. Again, nothing else. Yet show this structure to most organic chemists, and I venture to suggest few would immediately predict this (unless they are experts on benzidine rearrangements).‡

The eagle-eyed reader of this blog may have noticed my noting in previous posts that the benzidine rearrangement proper is normally promoted by double protonation, and that reaction via monoprotonation has a significantly higher barrier. So what are the corresponding predicted reaction barriers for PhNHOPh? I start in fact with catalytic monoprotonation. The calculations are at ωB97XD/6-311G(d,p)/SCRF=water (closed shell) level.
| System | N-protonated | O-Protonated‡ |
| Reactant | 0.0 | 11.3 |
| TS N-O | 7.3 | 17.4 |
| π-complex | 2.1 | 6.0 |
| TS C-C | 4.8 | 13.2 |
| ‡Relative to N-protonated reactant, in kcal/mol. | ||
So it seems that even monoprotonation (on nitrogen) results in a very small ΔG298‡ barrier to the formation of a π-complex and its subsequent facile breakdown to form a C-C bond. I had noted in the earlier post that Ghigo and co-workers[2] had found that with diprotonated diphenyl hydrazine, the resulting π-complex has some open shell (biradical) character. The calculations reported here on the monoprotonated system are done as closed shell, but any biradical character this might have will only serve to even further reduce the barriers seen in the table. So we may confidently conclude that even monoprotonated N,O-diphenyl hydroxylamine will rapidly rearrange. A follow-up investigation for the diprotonated route hardly seems necessary!
But here is a challenge: if one were able to prepare PhNHOPh in thoroughly deprotic conditions, might it be isolable? There is precedent; the keto form of phenol can indeed be isolated under such conditions.[3].
Here are some intrinsic reaction coordinates to finish with. Firstly, for the formation of the π-complex from N-protonated precursor:
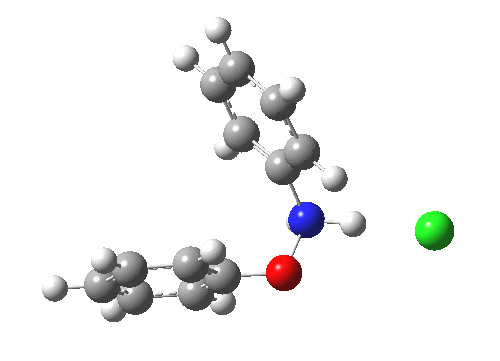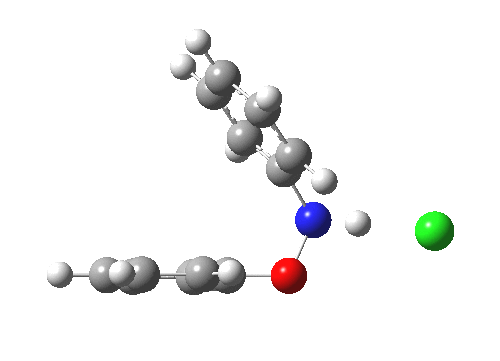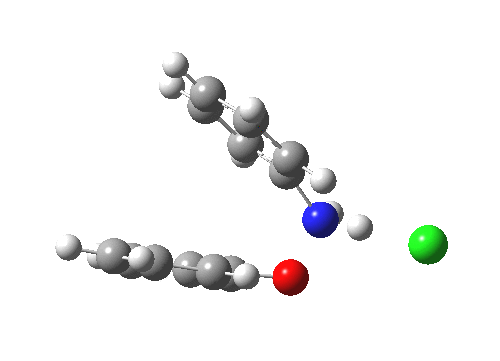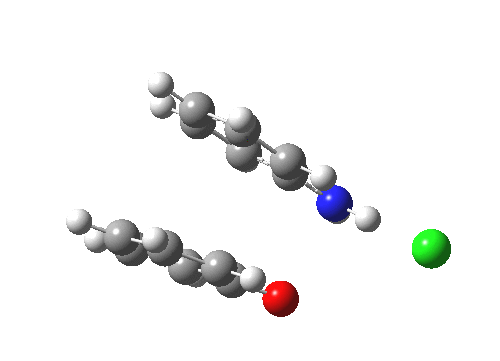 |
|
 |
 |
Once formed, the π-complex collapses readily to the 4,4′-coupled biphenyl.
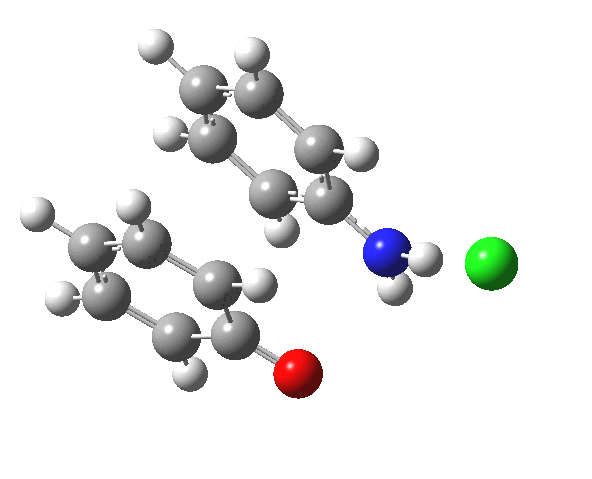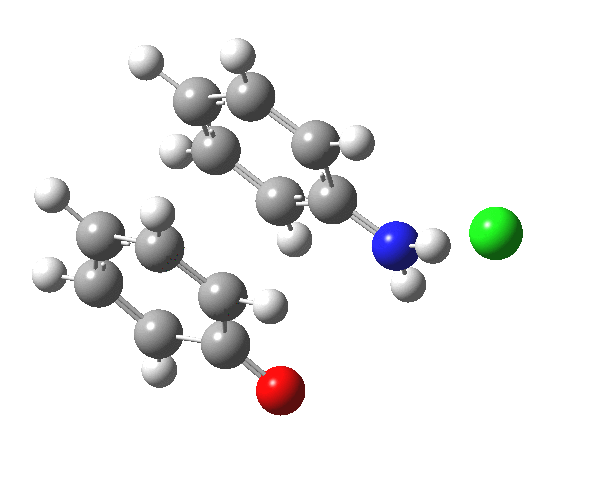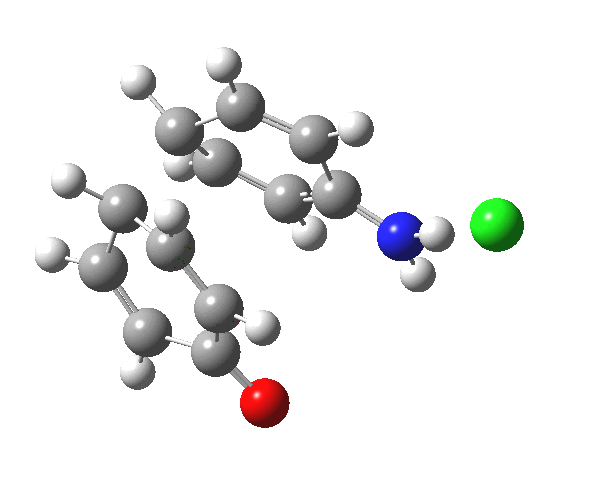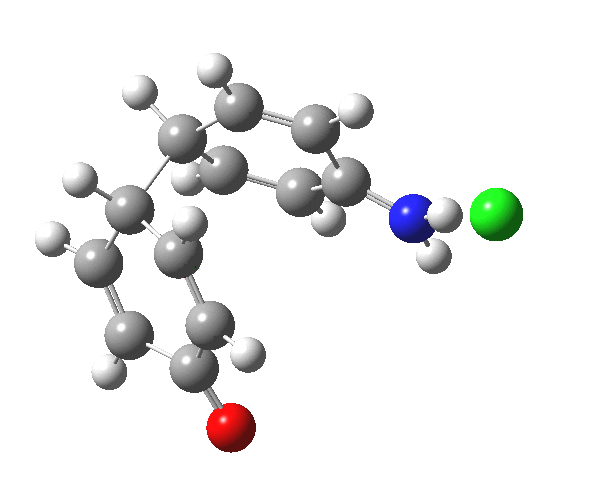 |
|
 |
 |
There may be another pathway which collapses to the 1,1′-coupled biphenyl which I have not found yet. A [3,3] sigmatropic rearrangement converting the 4,4′ to the 1,1′-biphenyl is higher in energy, but still just about accessible thermally.
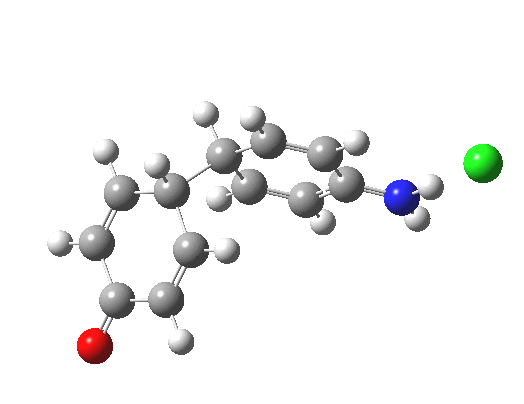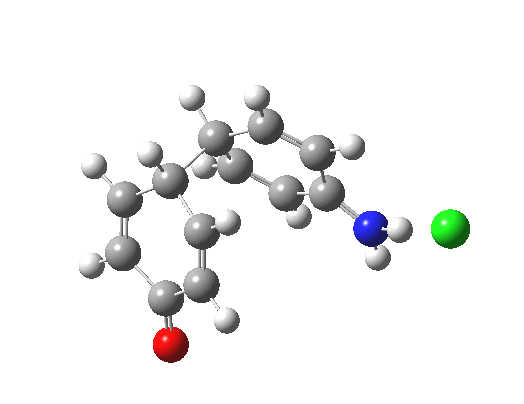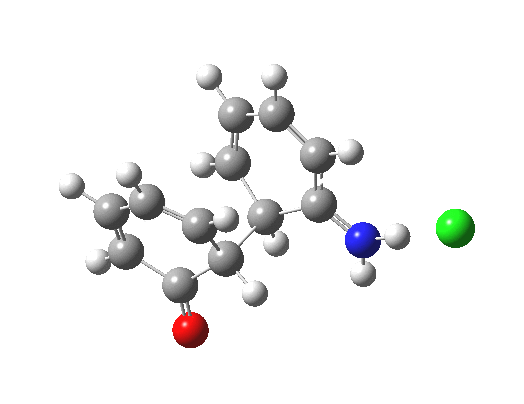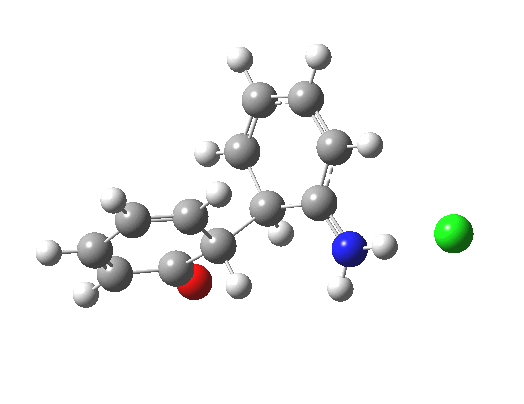 |
|
 |
 |
To end, here is a question. Could one systematically identify “gaps” in the distribution of known molecules; species which appear as if they should exist, but have never been reported? Of these, the majority will no doubt be absent from the record simply because they uninteresting. But some, as here, are absent because they are too unstable to exist, unless (extreme?) precautions are taken to remove the factors responsible for their instability (in this case, protons). Cyclobutadiene was one such famous example (stabilised by coordination to a metal). Certainly, computation nowadays can help identify conditions for how such molecules might be isolated.
‡In contrast, PhNHSPh (N-Phenylbenzenesulfenamide) is a well known species[4].
Author
References
- J.R. Cox, and M.F. Dunn, "The chemistry of O,N-diarylhydroxlamines - I", Tetrahedron Letters, vol. 4, pp. 985-989, 1963. https://doi.org/10.1016/s0040-4039(01)90757-9
- G. Ghigo, S. Osella, A. Maranzana, and G. Tonachini, "The Mechanism of the Acid‐Catalyzed Benzidine Rearrangement of Hydrazobenzene: A Theoretical Study", European Journal of Organic Chemistry, vol. 2011, pp. 2326-2333, 2011. https://doi.org/10.1002/ejoc.201001636
- B. Miller, "Preparation of the Ketone Tautomer of a Phenol by a Cope Rearrangement<sup>1</sup>", Journal of the American Chemical Society, vol. 87, pp. 5515-5516, 1965. https://doi.org/10.1021/ja00951a064
- I. Brito, A. Cárdenas, A. Mundaca, M. López-Rodríguez, and A. Reyes, "2-Iodo-<i>N</i>-(2-nitrophenylsulfanyl)aniline", Acta Crystallographica Section E Structure Reports Online, vol. 64, pp. o1387-o1387, 2008. https://doi.org/10.1107/s1600536808019491
Tags: energy, Historical, metal, pericyclic, Reaction Mechanism
[…] Chemistry with a twist « Why is N,O-diphenyl hydroxylamine (PhNHOPh) unknown? […]
Alex Genaev has emailed me this link to a script for concatenating .xyz files to produce an animation including more than one discrete IRC pathway. He used it to generate this image
In the same vein, I have never seen a diaryl peroxide reported, except maybe for ill-characterized stuff in one or two old patents. Has anyone else seen a well-characterized diaryl peroxide, where the O-O link was established beyond reasonable doubt, or a calculation on same?
Yes, REAXYS only has one citation, dealing with the kinetics of the formation of phenoxyl radicals and their coupling and there is nothing in CAS either. It is very likely that diphenyl peroxide rearranges via a benzidine-type reaction. Again, one may presume that rigorous removal of all acid might stabilize it.
Hydrazobenzene then is readily accessible, apparently since it is prepared “hydrochloride-free”.
Hydrazobenzene is stable because, unlike PhNHOPh, it only reacts rapidly if doubly-protonated. Since the concentration of the bis-protonated species is very low, the rate of reaction is likewise low. The mono-protonated mechanism (see here) has a much higher activation barrier than does PhNHOPh (or presumably PhOOPh).
[…] of diphenyl hydrazine. This reaction requires diprotonation to proceed readily, but we then discovered that replacing one NH by an O as in N,O-diphenyl hydroxylamine required only monoprotonation to […]