A few posts back, I explored the “benzidine rearrangement” of diphenyl hydrazine. This reaction requires diprotonation to proceed readily, but we then discovered that replacing one NH by an O as in N,O-diphenyl hydroxylamine required only monoprotonation to undergo an equivalent facile rearrangement. So replacing both NHs by O to form diphenyl peroxide (Ph-O-O-Ph) completes this homologous series. I had speculated that PhNHOPh might exist if all traces of catalytic acid were removed, but could the same be done to PhOOPh? Not if it continues the trend and requires no prior protonation at all!

Here is the results of a ωB97XD/6-311G(d,p)/SCRF=water calculation. Now I should explain that the conventional explanation for the non-existence of PhOOPh is that the O-O bond homolyses very readily to form phenoxy radicals[1]. But of course other peroxides such as t-Bu-O-O-t-Bu do exist (although they are rather fragile) and so the phenyl analogue is clearly special.
 |
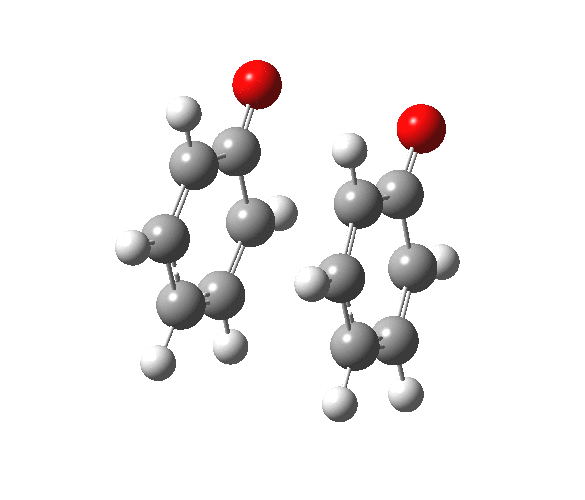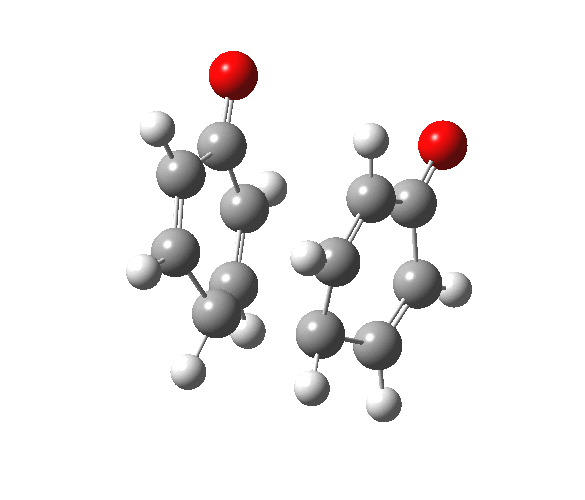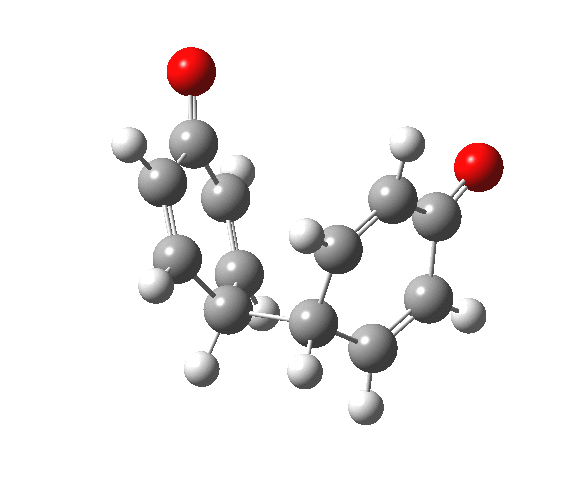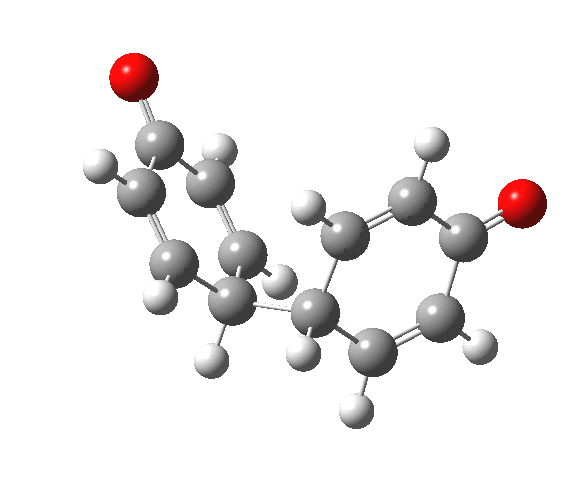 |
 |
 |
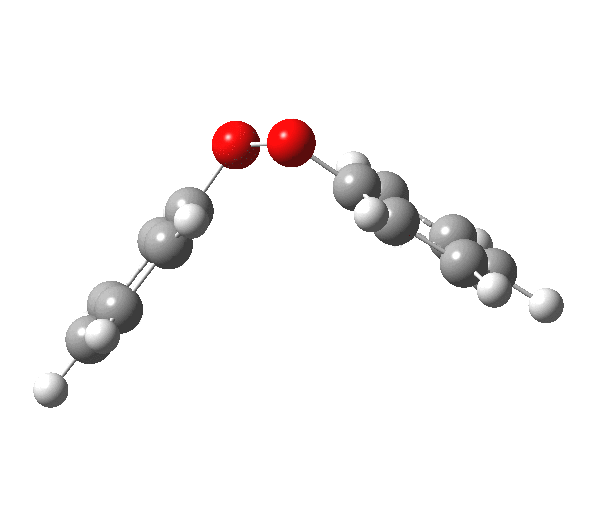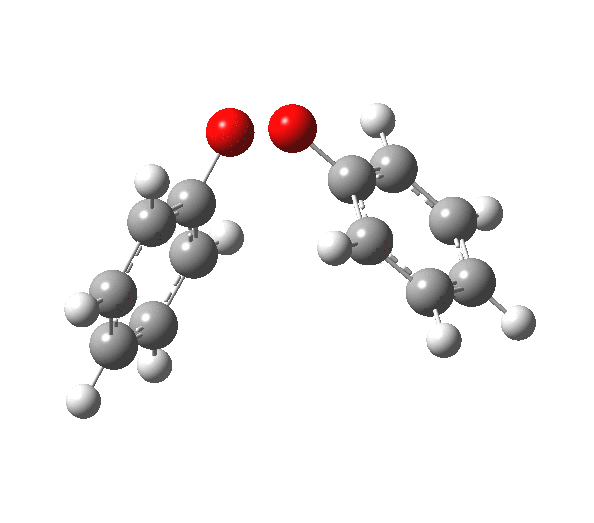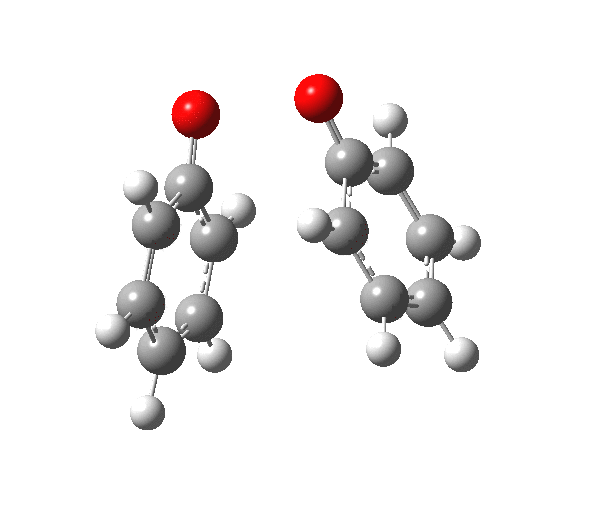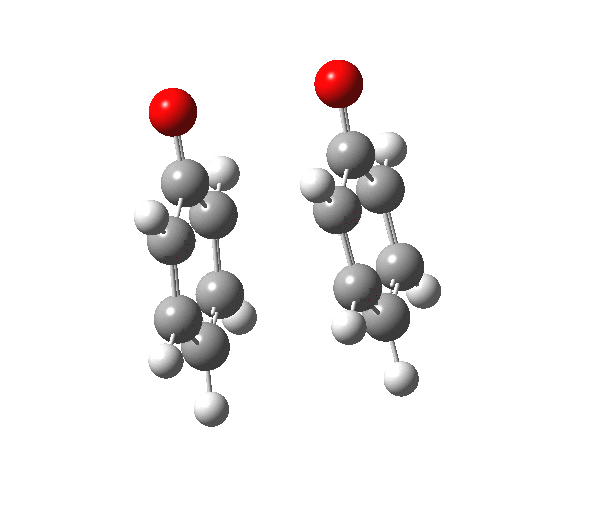
You will notice from the IRC profiles shown above that even without any prior protonation, the barrier to O-O cleavage is really very small (~ 4 kcal/mol). But the method I have used to calculate this is a closed shell DFT procedure. This does not allow the formation of the (open shell) biradical that two phenoxy radicals would represent. The barrier is low even without the formation of phenoxy radicals! Of course, as with the two previous examples, the actual initial product formed is the π-complex as first suggested by Michael Dewar. The wavefunction of such a species requires special treatment, since it is best described as a linear combination of two closed-shell configurations, what is called a multi-configuration or multi-reference wavefunction.‡ So the single-configuration closed shell calculation that the above IRC represents must be an upper bound to a proper description of the energy transition state. In other words, if the description is improved, the barrier can only get even lower!
Notice in the above that the π-complex formed in the first stage (of two) is actually lower in energy than the diphenyl peroxide itself, and that the barrier for this π-complex to then collapse to form the C-C bond between the two 4-positions is also tiny. This π-complex in other words is very transient indeed, probably not surviving for even one molecular vibration. To all intents and purposes, this really is a concerted [5,5] sigmatropic shift, as shown in the schematic at the top of this post. But the bottom line is that the homolysis argument need not be the only one (although it is not necessarily incorrect). One can just as readily explain why PhOOPh does not exist by invoking facile formation of Dewar-like π-complex instead.
‡ Another deceptively simple little molecule that requires such a treatment is C2, the topic of much recent debate![2], [3]
Author
References
- R. Benassi, U. Folli, S. Sbardellati, and F. Taddei, "Conformational properties and homolytic bond cleavage of organic peroxides. I. An empirical approach based upon molecular mechanics and <i>ab initio</i> calculations", Journal of Computational Chemistry, vol. 14, pp. 379-391, 1993. https://doi.org/10.1002/jcc.540140402
- S. Shaik, H.S. Rzepa, and R. Hoffmann, "One Molecule, Two Atoms, Three Views, Four Bonds?", Angewandte Chemie International Edition, vol. 52, pp. 3020-3033, 2013. https://doi.org/10.1002/anie.201208206
- J.M. Matxain, F. Ruipérez, I. Infante, X. Lopez, J.M. Ugalde, G. Merino, and M. Piris, "Communication: Chemical bonding in carbon dimer isovalent series from the natural orbital functional theory perspective", The Journal of Chemical Physics, vol. 138, 2013. https://doi.org/10.1063/1.4802585
Tags: actual initial product, energy, energy transition state, Michael Dewar, New Hampshire, Reaction Mechanism
Hi, professor Rzepa, what’s the meanig of horizontal coordinate in IRC?
Molecular Distance? Why can it be minus?
It is the integrated progress along the reaction coordinate, centred at the transition state, which is zero, and leading to either the reactant in one direction, or to the product in the other. The sign of the IRC is in effect random; the value has no great meaning.
Thanks very much for the clarification. Could you tell me where to get a short introduction about IRC? I didn’t find enough information on Wikipedia.
I have two organic books about mechanism, they often give different mechanism on the same reaction which makes me confused. Theory study is very helpful to clarify the real mechanism.
The above question prompts me to remind everyone of “CCL” (as it is known to its friends, or computational chemistry list“). It is a remarkable 22 years old now and thousands of questions such as the above have both been posted and often answered there. I suspect many of the current generation of computational chemists have learnt (at least in part) their trade there.
There is quite a lot there about IRCs, although much of it is how about to run one!