Eagle-eyed footnote readers might have spotted one at the bottom of the post on the benzidine rearrangement. I was comparing the N-N bond lengths in crystal structures of known diprotonated hydrazines (~1.45Å) with the computed N-N bond length at the start point of the intrinsic reaction coordinate for the [5,5] sigmatropic rearrangement of di-N-protonated diphenylhydrazine (the active species in the benzidine rearrangement itself), which was some 1Å longer. This post explores the implications of this oddity.

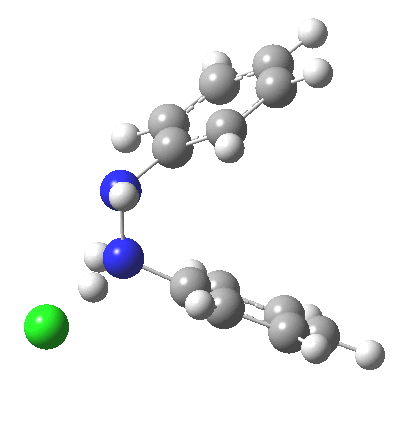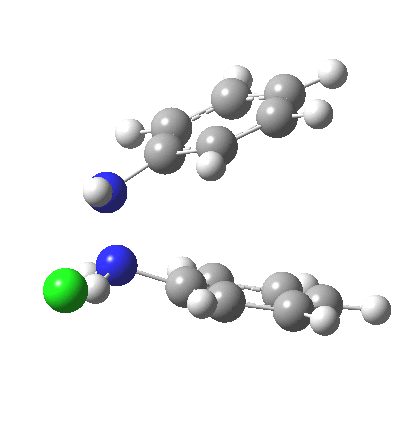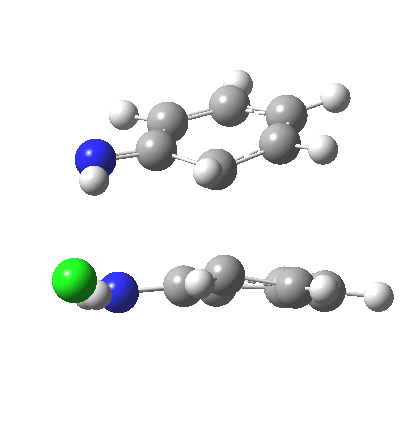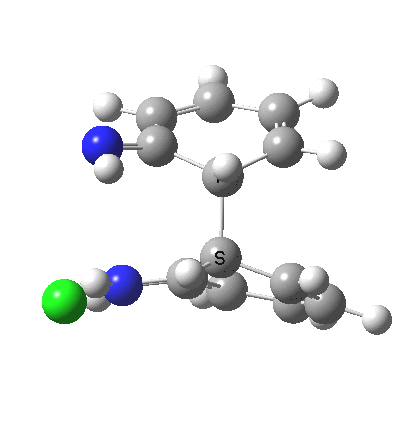
My start point however is actually the mono-N-protonated system. The IRC for the calculated transition state is shown below. The activation barrier is a lot higher than with the diprotonated route, but I want to bring to your attention a feature at IRC = +5 to +3. At this point the RMS gradient norm dips, approaching but not quite reaching zero. This is what is called a hidden intermediate, an intermediate that does not quite form.‡ It is in this region that the N-N bond length changes from the value of about 1.45Å for the monoprotonated hydrazine, to around 2.5Å at the point of the “hidden intermediate”. This represents the formation of the π-π-stacked complex as the preamble to the actual rearrangement, the transition state for which is of course reached at IRC =0.0. For this system, the [5,5] sigmatropic is actually slightly higher ( ΔG‡298 +2.4 kcal/mol) than the competing [3,3] rearrangement, which also shows that hidden intermediate ( at IRC ~+2.0). This close balance between the [3,3] and the [5,5] mechanisms suggests that factors such as ring substituents, counter-ion, solvent etc may in fact be able to swing this balance one way or the other.
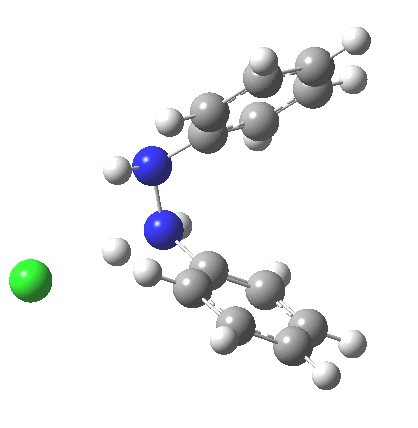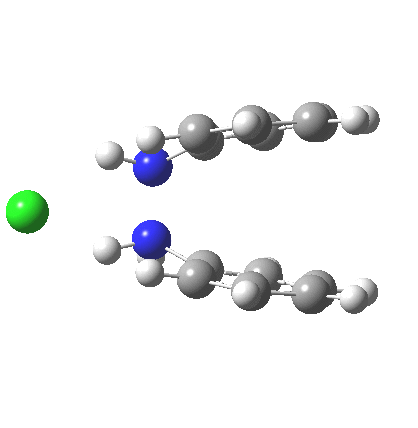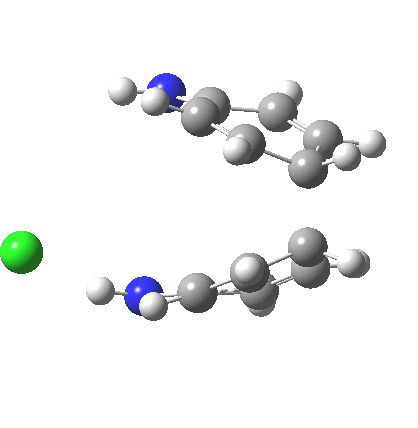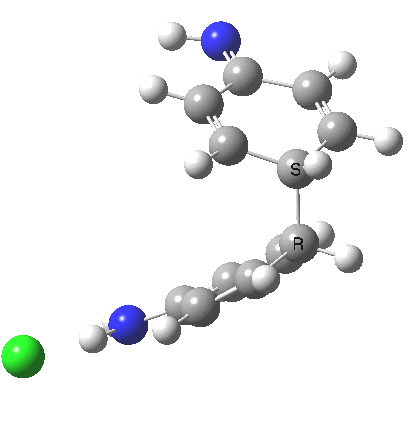 The 5,5, sigmatropic rearrangement of monoprotonated diphenylhydrazine. Click for 3D |
|
 |
 |
 The 3,3 sigmatropic rearrangement of monoprotonated diphenylhydrazine. Click for 3D. |
|
 |
 |
Which brings us back to the diprotonated species, the one with the N-N bond length of 2.53Å. This is a stable minimum (i.e. the RMS gradient norm is zero) with no imaginary frequencies computed, and hence it is no longer a hidden intermediate, but an exposed π-complex. Adding that second proton has stabilised it considerably. It is higher in ΔG298 than the anti-conformation of diprotonated diphenylhydrazine by 6.1 kcal/mol, the latter having the normal N-N bond length of 1.46Å. The free energy barrier from the π-complex to the transition state for [5,5] rearrangement (shown in previous post) is a mere 2.4 kcal/mol. The barrier from the same π-complex to the transition state (N-N length 1.97Å) leading back to N-N diprotonated diphenylhydrazine is also small, 3.1 kcal/mol, so this π-complex is bounded only by small barriers and hence is very unlikely to be directly detected.
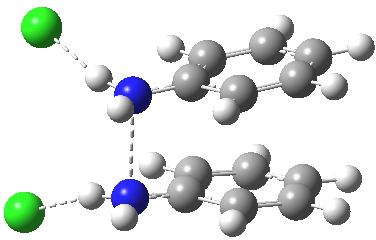 The benzidine π-complex. Click for 3D. |
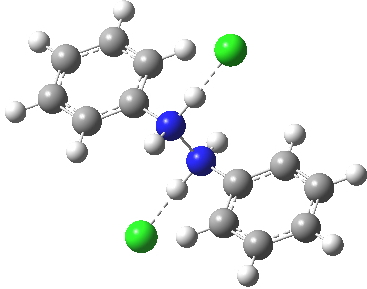 Anti-diprotonated diphenyl hydrazine. Click for 3D. |
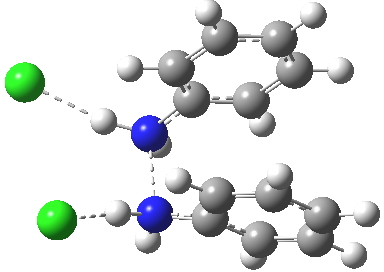 Transition state between π-complex and N-N diprotonated diphenyhydrazine. Click for 3D. |
|
To conclude, mono-protonated diphenyl hydrazine† rearranges to the 4,4′-diaminobiphenyl via the so-called benzidine rearrangement by a concerted process that involves a hidden π-complex forming before the transition state is reached. Diprotonation exposes this hidden complex formed from diphenylhydrazine. This complex is the true starting point for the [5,5] sigmatropic rearrangement (if it can still be called that). The overall reaction becomes more exothermic by in effect separating the two positive charges resulting from nitrogen diprotonation onto the two phenyl rings, an affect which also encourages the π-complex to form.
‡Another good example of such a species is the intermediate carbocation in the solvolysis of t-butyl chloride. This too is hidden.
†It is rather curious that Ph-NH-O-Ph is in effect unknown (apart from one patent). Could it be that it cannot be prevented from rearranging by the same mechanism as Ph-NH-NH-Ph?
[…] the range IRC ± (9 – 15), unexpected features appear (hidden intermediates if you check this post). A whole plethora of them. This is the conformational region where the methyl flags start waving […]
[…] benzidine rearrangement proper is normally promoted by double protonation, and that reaction via monoprotonation has a significantly higher barrier. So what are the corresponding predicted reaction barriers for […]
[…] with a twist « π-hydrogen bonds as a function of ring size. Hidden intermediates in the benzidine rearrangement. The monoprotonated mechanism. […]