Functionalisation of a (hetero)aromatic ring by selectively (directedly) removing protons using the metal lithium is a relative mechanistic newcomer, compared to the pantheon of knowledge on aromatic electrophilic substitution. Investigating the mechanism using quantum calculations poses some interesting challenges, ones I have not previously discussed on this blog.

My model will be the system above, based on the pyridine ring, and also carrying a directing group (R=Me, DG = O–). The reagent used to remove the hydrogen and to substitute it (with a carbon-metal bond) is an alkyl lithium. The arrow pushing I have shown is speculative, since at this stage we have no idea if it really is such a pericyclic process. Indeed things are about to get complicated when we find out that the structure of the electron deficient lithium alkyls is much more complex than one might imagine.
Fortunately, crystal structures are available. Let me start with n-butyl lithium, a very commonly used reagent[1]. This forms a complex cluster of six lithiums, in which each metal is surrounded by three CH2– terminii of the n-butyl anion, and vice-versa, each CH2– group is in contact with three lithium atoms (making the carbanionic carbon in effect hexa-coordinate).
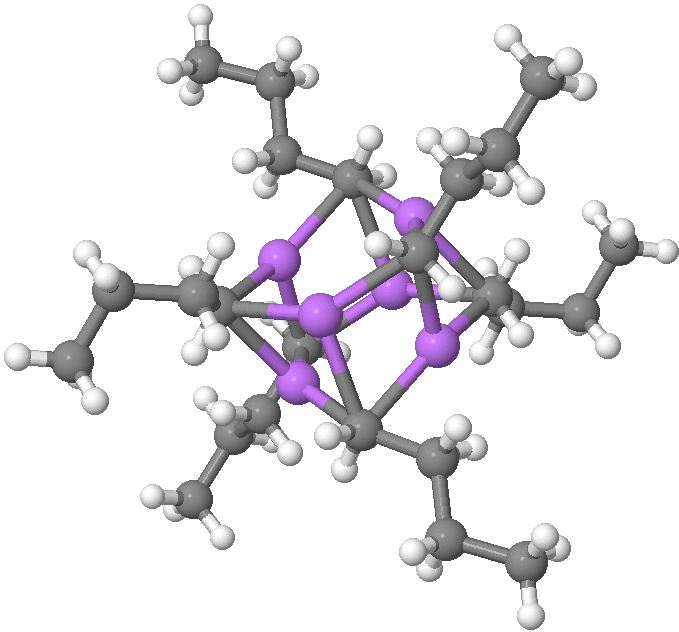
SUHBEC. CLICK FOR 3D.
Another frequently used lithium alkyl is the t-butyl derivative, which has a different tetrameric motif, again with each Me3C– coordinated to three Li atoms (making this carbon again hexa-coordinate).
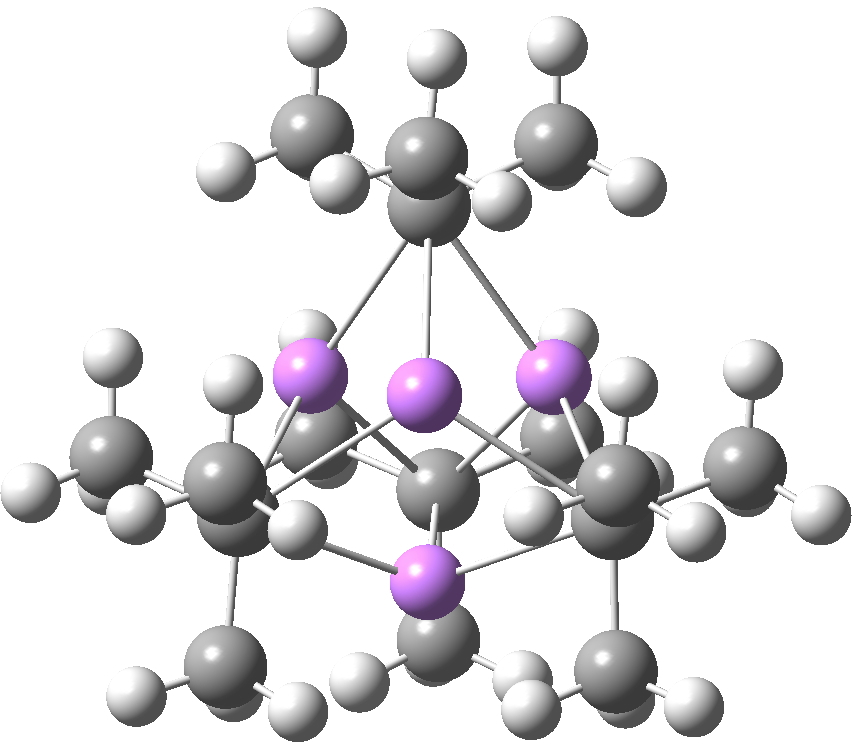
SUHBIG. Click for 3D.
The interesting issue now is whether these metal alkyls react in these oligomeric forms or whether they are in equilibrium with a reduced monomeric form that constitutes the reactive species. With n-butyl lithium, it is possible to try to achieve this chemically by adding tetramethylethylenediamine. As you can see from the structure below, this strategy can be only partially successful; in this instance the CH2– coordination is reduced from three Li atoms to two[2]. With t-butyl lithium, this strategy reduces the structure to a true monomer[3], the Me3C– now being just 4-coordinate.
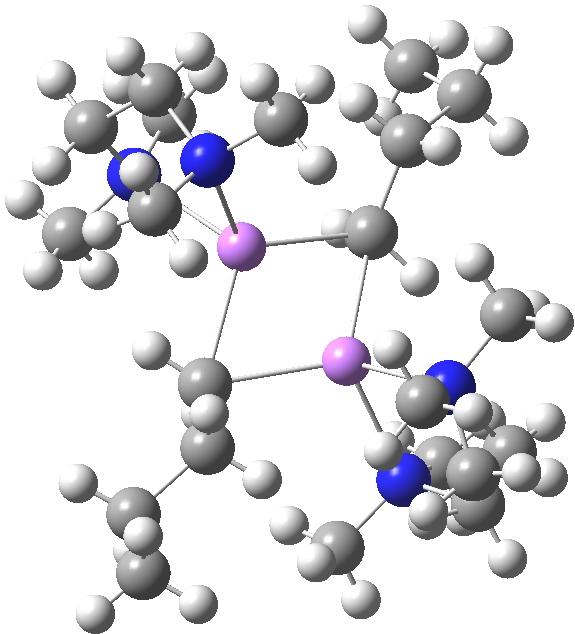 WAFJAO. Click for 3D. |
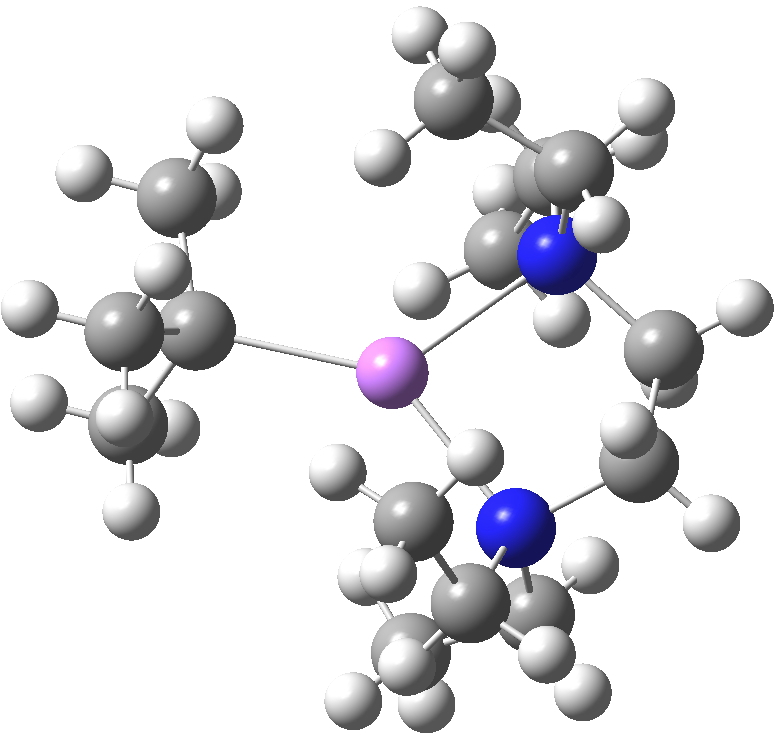 LOKTAH. Click for 3D. |
These systems are all pretty large to investigate using modelling, and so I will start the process by reducing the alkyl lithium model down to just a monomeric CH3Li molecule, placing it and pyridine-N-oxide into a continuum solvent cavity (ωB97XD/6-311G(d,p)/SCRF=benzene) and seeing what happens[4]. You can see it is both facile and a concerted process, corresponding pretty much to the arrow pushing illustrated at the top of this post.
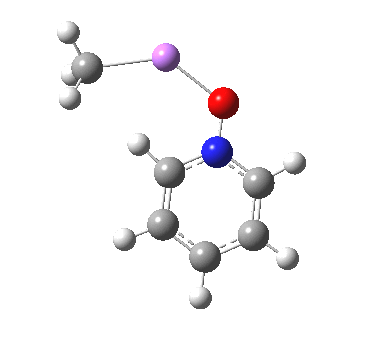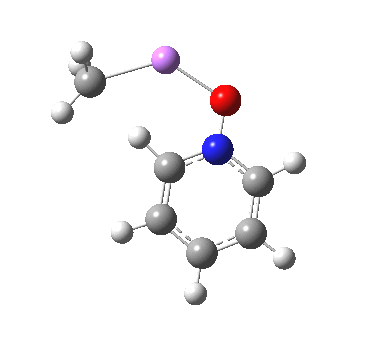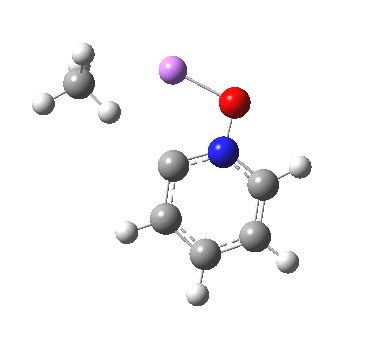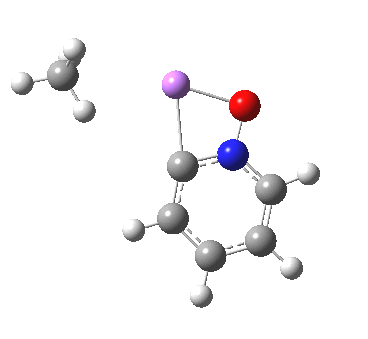 |
 |
But wait, where have we seen an aromatic substitution reaction which does exactly this in a single concerted step, first remove a proton and then replace it with an electrophile? This was in fact revealed in the IRC for electrophilic substitution of indole in the 1-position! Of course, there is a difference. With indole, we had pseudo-inversion at the nitrogen centre (a pseudo-Sn2 reaction if you will), whereas here it is pseudo-retention at the 2-carbon.
Is this model robust? Let us try a dimeric (MeLi)2 model coordinated to one pyridine-N-oxide. The IRC[5] is very similar, but the initial barrier to proton transfer is lower.
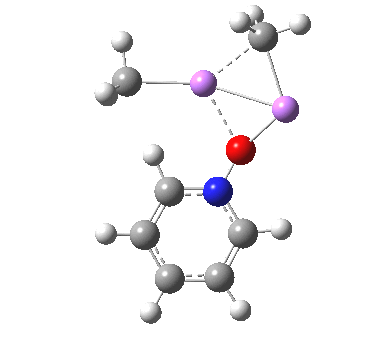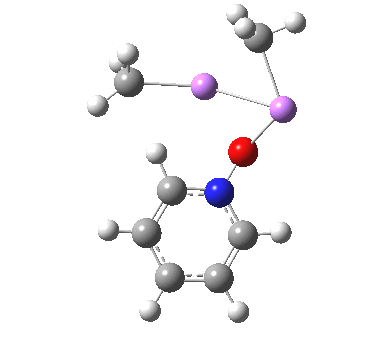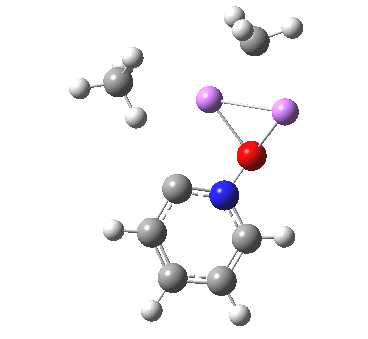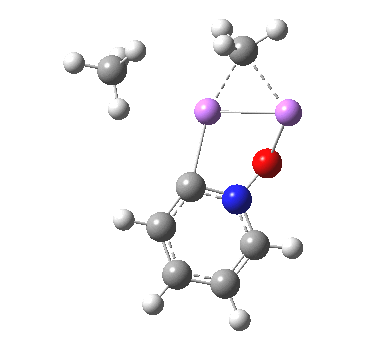 |
 |
Next, we have a model in which two molecules of pyridine-N-oxide (PNO) aggregate around two molecules of MeLi. This model is starting to resemble the tetramethylethylenediamine partially de-aggregated n-butyl lithium structure shown as WAFJAO above. The basic features[6] of the process remain intact, including the small barrier.
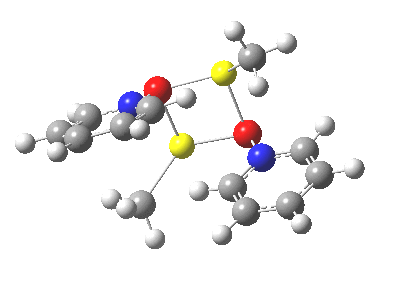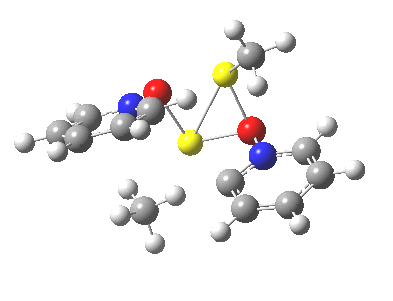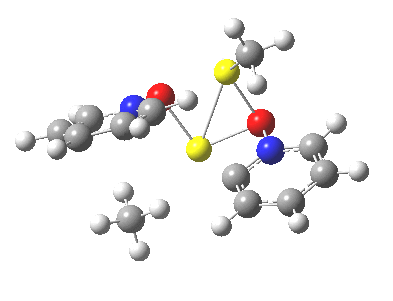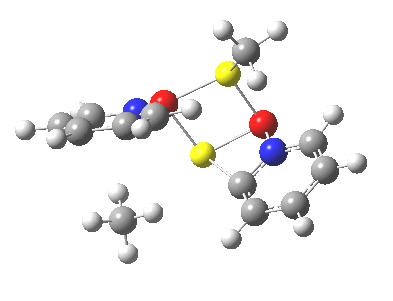 |
 |
Finally, I go back to the simple model, but with the directing group (DG) removed to give just pyridine. The profile[7] is the same, but the barrier is much larger. So perhaps both aggregation and coordination to a directing group help accelerate the reaction?
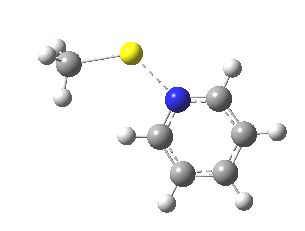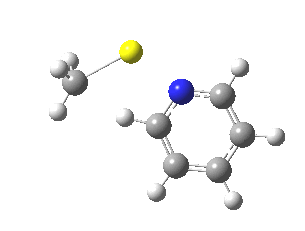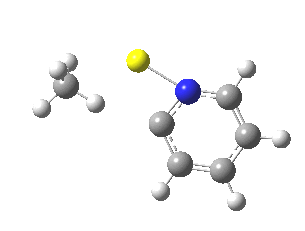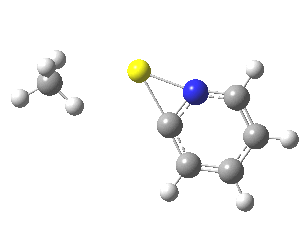 |
 |
So two reaction types, not normally associated with each other, turn out to have some intriguing similarities and an interesting difference.
Author
References
- T. Kottke, and D. Stalke, "Structures of Classical Reagents in Chemical Synthesis: (<i>n</i>BuLi)<sub>6</sub>, (<i>t</i>BuLi)<sub>4</sub>, and the Metastable (<i>t</i>BuLi · Et<sub>2</sub>O)<sub>2</sub>", Angewandte Chemie International Edition in English, vol. 32, pp. 580-582, 1993. https://doi.org/10.1002/anie.199305801
- M.A. Nichols, and P.G. Williard, "Solid-state structures of n-butyllithium-TMEDA, -THF, and -DME complexes", Journal of the American Chemical Society, vol. 115, pp. 1568-1572, 1993. https://doi.org/10.1021/ja00057a050
- V.H. Gessner, and C. Strohmann, "Lithiation of TMEDA and its Higher Homologous TEEDA: Understanding Observed α- and β-Deprotonation", Journal of the American Chemical Society, vol. 130, pp. 14412-14413, 2008. https://doi.org/10.1021/ja8058205
- H.S. Rzepa, "Gaussian Job Archive for C6H8LiNO", 2013. https://doi.org/10.6084/m9.figshare.651068
- H.S. Rzepa, "Gaussian Job Archive for C7H11Li2NO", figshare, 2013. https://doi.org/10.6084/m9.figshare.651764
- H.S. Rzepa, "Gaussian Job Archive for C6H8LiN", 2013. https://doi.org/10.6084/m9.figshare.653672
Tags: carbon-metal bond, lithiation, metal, metal alkyls, metal lithium, pericyclic, Reaction Mechanism, Tutorial material
[…] Chemistry with a twist « Lithiation of heteroaromatic rings: analogy to electrophilic substitution? […]
[…] two previous explorations of aromatic substitutions have involved an electrophile (NO+ or Li+). Time now to look at a nucleophile, representing nucleophilic aromatic substitution. The mechanism […]