Sometimes, connections between different areas of chemistry just pop out (without the help of semantic web tools, this is called serendipity). So here, I will try to join up some threads which emerge from previous posts.
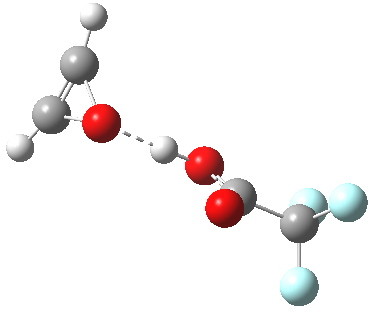
The original alkyne+peracid study was conducted using a gas phase model. I decided to revisit it now, but to change the modelled medium from the gas phase to continuum water. I show the IRC (intrinsic reaction coordinates) for this reaction in continuum water followed by the gas phase below (click on the animations to see the transition state model).
I want to compare the difference that introducing a model solvent (water) has made to the appearance of the reaction path.
The take home message is that the very nature of a reaction, the geometry (symmetry) of the molecules taking part, and the timing of the changes can be very visibly changed by simulating the event with a solvent. In the past of course, all such computational studies were conducted purely as a gas phase model.
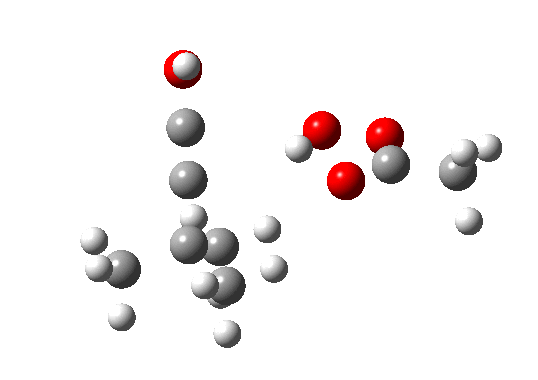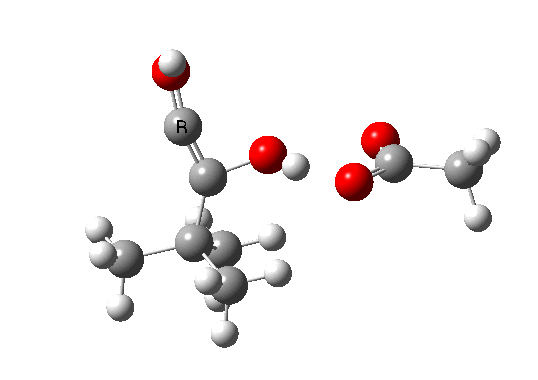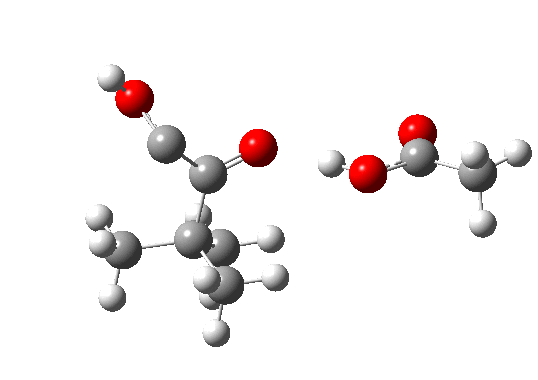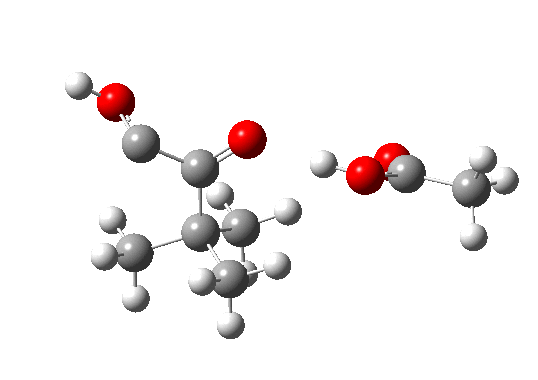
Postscript: The above shows how even a change in continuum solvent can affect the features of the reaction path. A rather greater perturbation is to change e.g. the substituents on the alkyne. I have tried replacing one H with t-butyl, and the other with OH. The rationale for the former is that t-butyl acetylene is actually the substrate that this reaction has been performed on, and for OH that it pushes electrons into the oxirene, making it more anti-aromatic and hence more liable to avoid that antiaromaticity. Animation of the IRC for this combination is shown below. Notice how the reaction now proceeds in a concerted manner directly from the alkyne to the hydroxy-carbene, without any sign of an intervening oxirene.
The energy and gradient profiles for this variation are shown below. Notice in particular how the barrier has dropped; it is now a much easier reaction.
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
View Comments
This example nicely illustrates a very important point that using only gas-phase calculations or making only single-point corrections for solvation free energy may be highly misleading. Things are just too different in vacuum: intermolecular interactions are exaggerated, charge separation is hindered, etc. So, the potential energy surface may have very different profile, and even the wavefunction character may change: I've seen open-shell singlets, stable in the gas phase, collapsed in continuum solvent.
I begin to think that if the system one works on is in solution, everything should be done with continuum solvent model applied. Fortunately, unlike not too long ago, there are some very robust implementations of continuum models available out there. Warts and all, it's still better than doing calculations in vacuum.
This of course brings a contentious issue of doing vibrational frequency calculations with PCM-type models and using them to get thermodynamic data, a truly heretic idea for some researchers. Although I think I do understand their logic, form the experience we've seen many times that it works fine.
Indeed, for species that don't change much in solution the frequencies and thermodynamic values hardly differ in the gas phase and in solution. In fact, a recent paper from Cramer and Truhlar groups have shown just that: DOI:10.1021/jp205508z