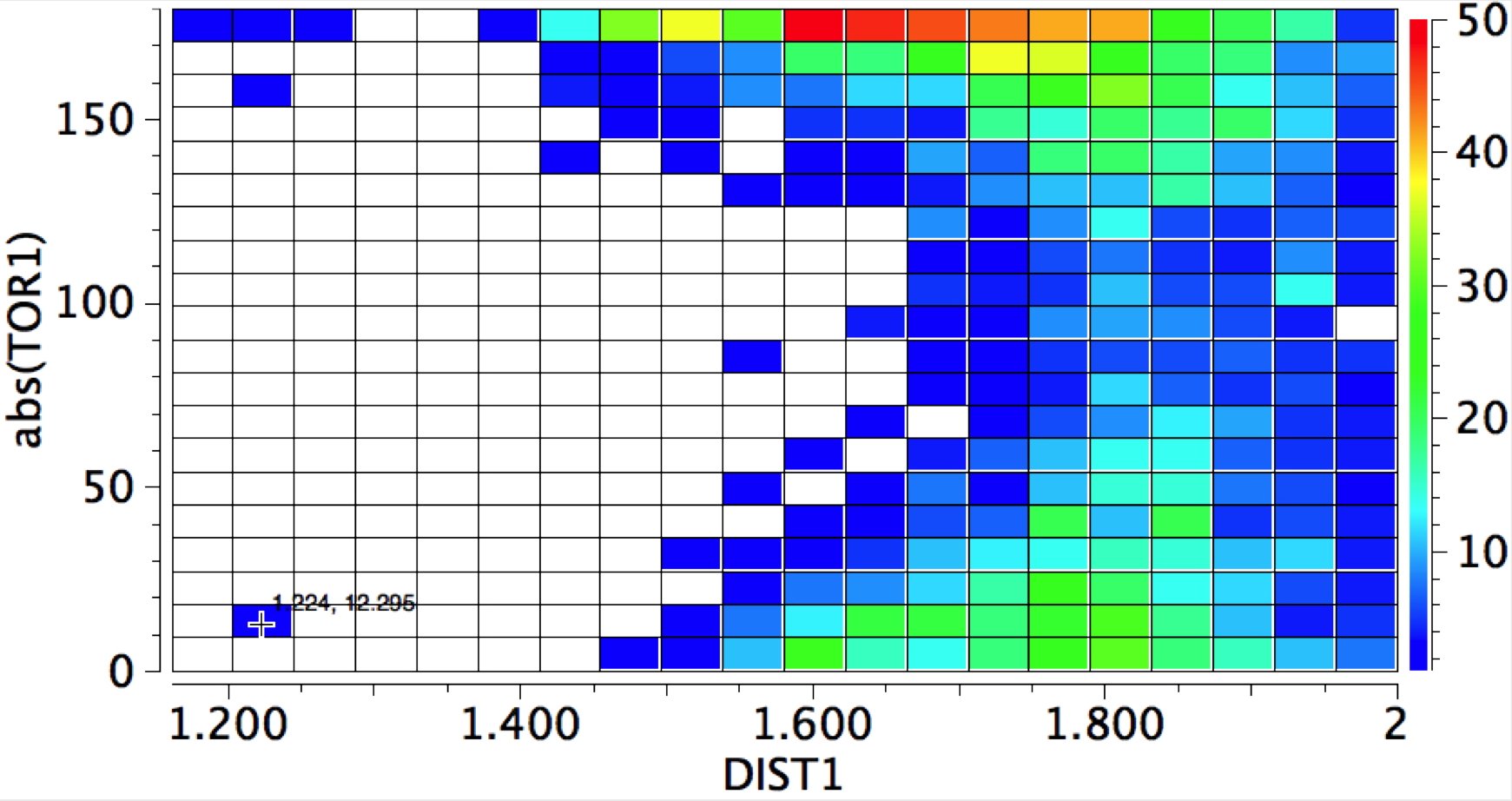
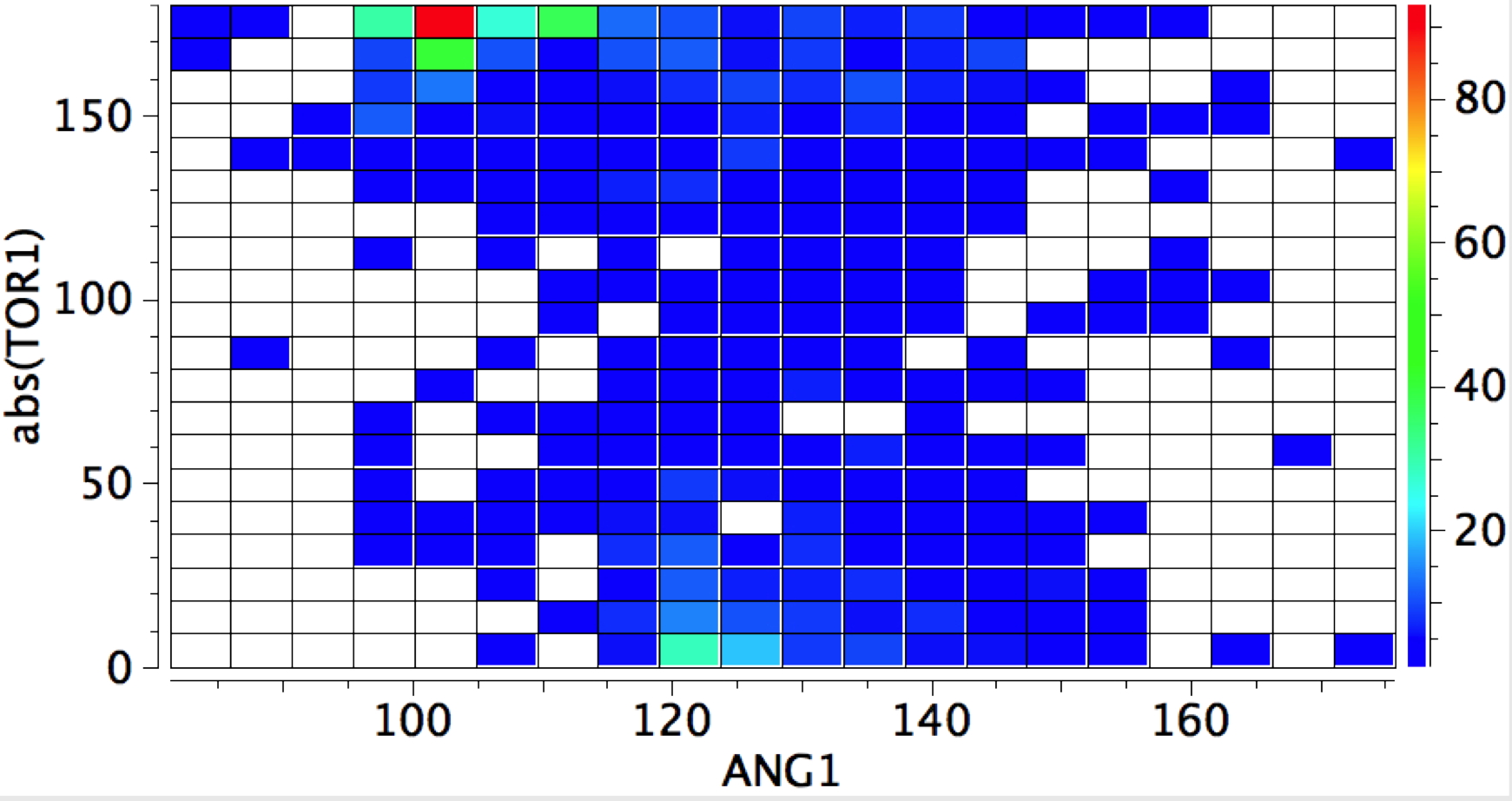
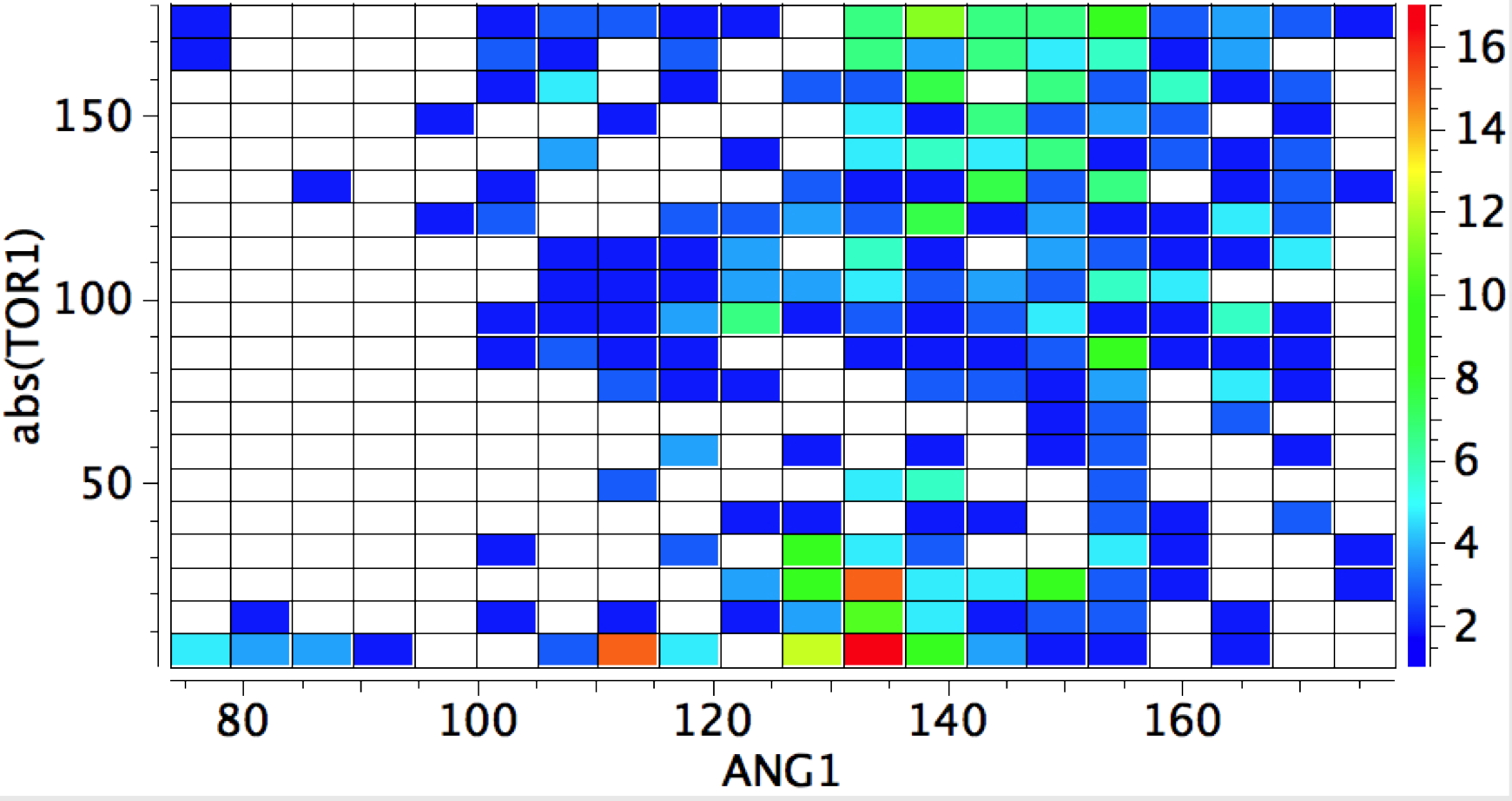
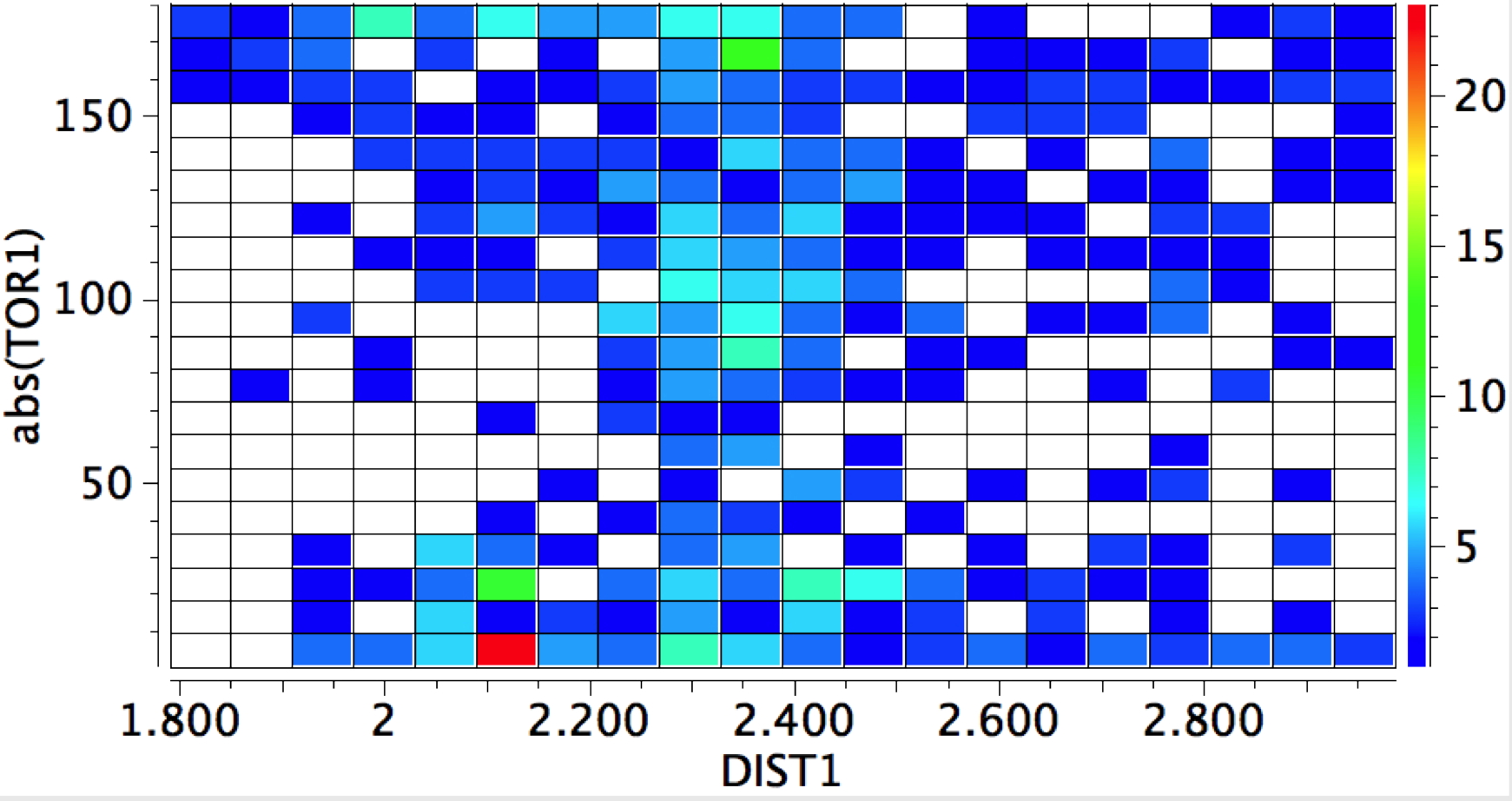
The Bürgi–Dunitz angle describes the trajectory of an approaching nucleophile towards the carbon atom of a carbonyl group. A colleague recently came to my office to ask about the inverse, that is what angle would an electrophile approach (an amide)? Thus it might approach either syn or anti with respect to the nitrogen, which is a feature not found with nucleophilic attack.
The above is a very general statistical survey. As with most bonding effects, one really should investigate every example to discover any perturbing circumstances or structural motifs that might distort the outcome. But for a ten minute exercise in response to a fascinating question from a colleague, it's not bad! And it certainly nicely inverts the usual Bürgi–Dunitz view of carbonyl groups.
In an earlier post, I discussed a phenomenon known as the "anomeric effect" exhibited by…
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}