The potential energy surface for a molecule tells us about how it might react. These surfaces have been charted for thousands of reactions using quantum mechanics, and their basic features are thought to be well understood. Coming across an entirely new feature is rare. So what do you make of the following?
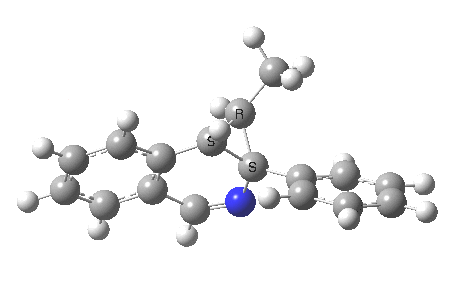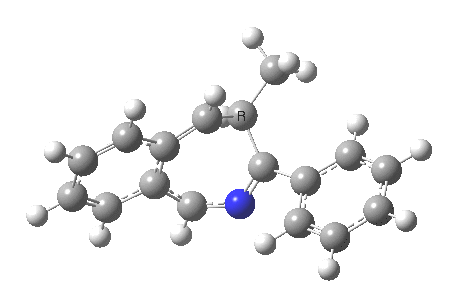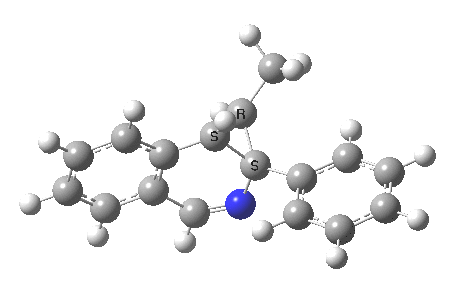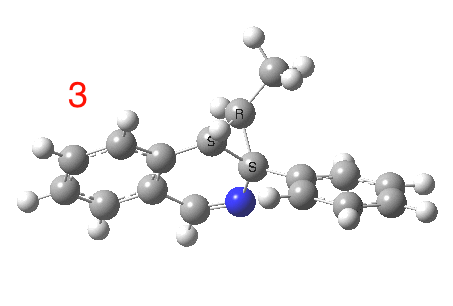
The reaction is shown above[1], and on the face of it, it looks like a normal pericyclic cascade. The standard mechanism inferred from simple mechanistic rules is to rewrite the ylid 1 as a carbene 2. This then undergoes a carbene insertion into an alkene to give 3, followed by an electrocyclic ring opening to give a presumed intermediate 4, and finally a [1,5] hydrogen migration ending in 5. Fairly uncontroversial stuff, you might think. The criterion is that it looks reasonable (each step has precedent).
The above reaction was discovered in 1992, before such simple mechanistic speculation could really be followed up by a good quality quantum mechanical investigation of how reasonable it really was. Well, this is 2014, and one need really spend no more than a few hours finding out. Before I present the results, it is worth reminding of the basic features of a potential energy surface:
- Reactant (intermediates) and products are all minima in such a surface. They are characterised by having 3N-6 (N= number of atoms) +ve force constants, and all the first derivatives (of geometric variables with respect to energy) are zero, as they also are for the next three types.
- A pair of minima can be connected by a transition state (a first order saddle point), for which 3N-7 of the force constants are +ve and precisely one is -ve.
- Less useful for mechanism are the 2nd order saddle points, which often connect a pair of transition states, and these have 3N-8 +ve force constants and 2 -ve ones. They are not kinetically important.
- Rarely, one finds two first order saddle points connected by a so-called valley ridge, and so one transition state can go downhill to another, and thence bifurcate into two possible products via a valley-ridge inflexion point.
- These four basic features have recently been augmented by so-called hidden intermediates. These emerge as a feature on the intrinsic reaction coordinate (IRC), being a frustrated minimum along that pathway. Frustrated, because the first derivatives never quite become zero (and the energy never quite a minimum) and so it does not qualify for any of the above definitions. Such points are increasingly being used to infer how small design changes to the reacting molecule might either fully stabilize such an intermediate, or perhaps remove it.
What we are about to discover relates to category 5, but with a new twist. Let us start with the IRC for 2 → 3[2] computed as ωB97XD/6-311G(d,p)/methanol.
- The first noteworthy feature is that the activation energy for this reaction is tiny (~ 2 kcal/mol). The reactant 1 is in fact generated in situ from the imidoyl chloride and potassium t-butoxide; in effect it reacts as soon as it is formed!
- After this early transition state has passed, the reaction appears to pause at IRC ~3.6. This is a nice example of a hidden intermediate. You can see from the gradient plot of the IRC that the derivatives became small at this point, but do not quite become zero. I have set the relative energy to be zero at this point, for a reason which will soon become apparent. And then the reaction picks up again.
- In the IRC region 8-14, we get a conformational phenomenon, the slow rotation of the phenyl group.
So, the scheme at the top is not correct! Species 4 is reached BEFORE species 3, and 4 is a hidden rather than a real intermediate.
So to the next reaction, which is the [1,5] hydrogen shift.[3] This in fact starts where the last left off, at 3, and ends at 5. But again, 4 crops up as a hidden intermediate! It is common to BOTH reactions.‡ Two completely different types of reaction share 4 as a common hidden intermediate. Think of it as two flight paths intersecting at a common point in 3D space.
- This reaction can be thought of as a concerted pericyclic cascade. By this I mean two consecutive pericyclic processes, separated not so much by a real intermediate as a hidden one (4). A conjoined pericyclic if you will.
- A reality check. The original report says that the reaction of 2 → 3 occurs at 25°C, and 3 is fully characterised by NMR. The next phase, 3 → 5 only occurs at 70°C. The rate at which 2 → 3 forms must be determined by the rate of formation of 1/2 and not by the pathway shown above. Then the route for 3 → 5 crosses the route taken by 2 → 3 and proceeds on upwards to the transition state for [1,5] H transfer. One might argue that when the 3 → 5 journey has reached 4 it has two options; to continue on to 5, or to go on to 2. Another question might relate to the original journey of 2 → 3. When it reaches point 4, could it then take a sharp turn and instead head for 5 thus by-passing 3 entirely? Well no, because at this stage 3 is entirely downhill, where 5 needs some more climbing doing.
- The reality check has just one fly in the ointment; the barrier to the [1,5] shift is ~40 kcal/mol, about 15 kcal/mol too high to occur at 70C. It might be that instead we have a base-catalysed bimolecular deprotonation/reprotonation as a competing pathway.
Nonetheless, it would be interesting to act as an observer and stand at crossroads 4 watching molecules go by. Some are coming from 2 and headed for 3, some are coming from the other direction heading for 5. Each set has a sense of direction of where they are headed (and memory of where they have come from). You might spot where I am going with this; molecular dynamics! But 4 certainly is an interesting feature on the potential energy surface of this system, and not one I have ever seen before (indeed, has anyone seen similar?).
‡ Because 4 is not a stationary point in the potential surface (its gradients are not zero), it can only be characterised in the context of the IRC pathway. So its two manifestations in the two different IRCs are very similar, but are not identical.
References
-
K.R. Motion, I.R. Robertson, J.T. Sharp, and M.D. Walkinshaw, "Reactions of diene-conjugated 1,3-dipolar intermediates: the formation of cyclopropa[c]isoquinolines from benzonitrile o-alkenylbenzyl ylides and their rearrangements to benzazepines", Journal of the Chemical Society, Perkin Transactions 1, pp. 1709, 1992. http://dx.doi.org/10.1039/P19920001709
-
Henry S. Rzepa., "Gaussian Job Archive for C17H15N", 2014. http://dx.doi.org/10.6084/m9.figshare.936551
-
Henry S. Rzepa., "Gaussian Job Archive for C17H15N", 2014. http://dx.doi.org/10.6084/m9.figshare.936657
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
View Comments
Tobias: "Maybe exposing such content on a much wider scale, such as open electronic textbooks (http://chemwiki.ucdavis.edu/) "
I took a very quick look at chemwiki.ucdavis.edu/
Organic_Chemistry/Pericyclic_Reactions/Cycloaddition_Reactions which is the equivalent in terms of content of 10042/a3uy9. It is conventional in the sense that it could easily be a PDF file, and it does not contain any animations, transition states, orbitals or other 3D content.
Our own Wiki (also named ChemWiki when we set it up in March 2006) was from the outset enabled to show 3D content, and we use it still to teach various aspects of molecular modelling. It will shortly be refactored to also support tablets (they were not around in 2006!). And it has always been open!
I might finally add that we too are discussing MOOCs here. I think the phrase is distinctive contributions to the genre. Thus if you compare my take on pericyclic reactions and Bill Reusch's, I think you will find them very very different. Dare I say, each is distinctive! I would not want the MOOC phenomenon to end up with the concept that each topic can/should be covered once in a global sense, and then all students are sent off to that one instance to "learn" about any particular topic. Diversity is good for science and good for chemistry. Pedagogy is all about assimilating the diversity and drawing your own conclusions from it. In that sense, I am not warming to MOOCS; they may simply homogenise, remove diversity, and cheapen everything. But those of us not charged with being the custodians of say the global MOOC on pericyclic reactions must redouble our efforts to be distinctive.
I set 10042/a3uy9 up very much with that in mind.