organic chemistry. It does not look like much, but this small little molecule brought us ferrocene, fluxional NMR, aromatic anions and valley-ridge inflexion points. You might not have heard of this last one, but in fact I mentioned the phenomenon in my post on nitrosobenzene. As for being at a crossroads, more like a Y-junction. Let me explain why.

Cyclopentadiene is made by thermal cracking of its dimer, and on standing it slowly reverts to this species. At its simplest, this dimerisation can be described as a π2s + π4s pericyclic cycloaddition, one of the monomers being the π2s and the other the π4s. Two new bonds are formed; one of these is shown in black, but the other can be either the one in red (which makes the π4s the monomer on the right) or the one in blue (in which case the π4s comes from the molecule on the left). How do these two partners decide which role each is to play? Well, the short answer is that, initially at least, they do not! The reaction proceeds very asynchronously, forming at first only the black bond. Eventually, they cannot take the suspense any longer, and when the point indicated with a green dot is reached, they finally have to take a decision. Up to the green dot, the potential energy surface has followed along a valley ridge, and the green decision point is known as the bifurcation point; one with an equal probability of the reaction giving either the top dimer or the bottom dimer.

If you are sharp-eyed you may notice a methyl group has been added to one of the monomers; this was done to balance the decision very slightly in favour of one route down from the green point over the other. Otherwise, the IRC pathway often just stops at the green point, unable to decide which way to take.


You can see this oddity reflected in the gradient norm of the IRC, which at IRC -1.5 suddenly acquires a new feature, the formation of the second bond. The lesson here is to remember that bonds do not have to form at the same time, they can instead follow, one after the other.
The two different dimers that result from the bifurcation are not in fact identical, they are mirror images (diastereomers because of the methyl group) of each other. They can in turn be inter-converted by a Cope rearrangement, a [3,3] sigmatropic reaction. The transition state for this process is none other than the green point reached earlier. It is indeed a transition state at a crossroads, connecting two quite different reactions, the Diels-Alder cycloaddition and the [3,3] Cope enantiomerisation of the dimer product. Such a reaction has been christened a bispericyclic reaction, one truly at a Y-junction.
Who would have thought that such an un-pretentious molecule could teach us so much. You can see this and many other examples of pericylic reactions in my course on the topic, available on an iPad by clicking here.
Postscript: I have managed to run a full IRC on the system without the methyl perturbation.

The bifurcation point (green dot) is clearly seen in the following two plots at a value of IRC +1.0
 |
 |
Author
Tags: dimer product, iPad, pericyclic, Postscript, potential energy surface, Reaction Mechanism
CPD itself also brought us rapid 1,5-H shifts as well as hyperconjugative aromaticity. Mulliken realized long ago that its CH2 group could function as a 2 π electron donor. The readily available CPD dimer, dicyclopentadiene (DCPD), gives adamantane in two steps.
The CPD dimerization led to the Alder-Stein rule of endo-addition (unfotunately, Henry depicts DCPD with the incorrect exo stereochemistry in the two right side representations). The role of secondary orbital interactions in determining the correct endo stereochemistry is not brought out explicitly, but should be apparent from this post. Endo (but not exo) DCPD undergoes the degenerate Cope rearrangement.
Thanks Paul for the insights. The rapid [1,5] shifts were first discovered, I think, when its 1H NMR spectrum was first measured, probably in the mid to late 1950s. I know that Geoff Wilkinson (who was interested in it in terms of the ferrocenes) had asked that an early low temperature probe be constructed on one of the first commercial NMR systems (40 MHz?) round about that period in order to show that the spectrum became more complex as you cooled it. This is what I alluded to in my reference to fluxional NMR behaviour. Whether he was the first to do this remains unknown. I have however speculated that if he HAD observed the behaviour, and rationalised it as a rapid [1,5] shift, and then attempted to explain it theoretically, he would have anticipated Woodward by 5-6 years! But he did not!
Regarding the endo stereochemistry on the rhs, I actually did not show any stereochemistry at the bridgehead (the implicit hydrogens need a wedge). I was trying to depict how a [3,3] sigmatropic shift converts the dimer into its mirror image; this of course preserves the endo relationship of the rings throughout.
It has been argued that this particular system is the one that best illustrates the secondary orbital interactions, via the ambiguity of which monomer acts as a diene and which as the alkene.
I am adding here another IRC animation. This time it starts from the green dot, itself of course a transition state, and tracks the [3,3] sigmatropic shift that occurs. The endo nature of both start and end dimers can be seen.
[…] of path known as a valley-ridge inflection path. An example of such a surface can be seen for the dimerisation of cyclopentadiene (DOI: 10.1021/ja016622h) and effectively it connects one transition state to a second transition […]
The results in this post relate to what is called the endo geometry of the cycloaddition. This is the normal preferred mode for many cycloadditions. To this transition state (doi: 10.6084/m9.figshare.865607) I have now computed the energy of the exo-transition state (doi: 10.6084/m9.figshare.865610), which emerges as 3.3 kcal/mol higher than the endo isomer (as is observed from the stereochemistry of the product formed).
Here is an unusual way of representing the secondary orbital interactions which are normally the explanation attached to the endo-preference for cycloadditions. This is an NCI (non-covalent-interactions) analysis.
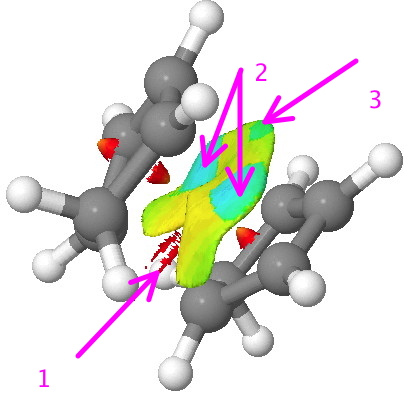
Arrow 1 points to the forming short bond (most of the NCI is filtered out by the threshold applied so as to conceal covalent normal bonds). Arrow 2 points to the two other bond forming regions, only one of which actually ends up forming a bond, depending on which way the bifurcation at the valley-ridge inflexion point goes. The blue indicates this region is already strongly stabilising (a precursor to forming a bond). Arrow 3 points to another point of stabilisation, but this one does not end up forming a bond. These three blue regions can be interpreted as the stabilisations resulting from those secondary orbital overlaps.