The Baeyer-Villiger rearrangement was named after its discoverers, who in 1899 described the transformation of menthone into the corresponding lactone using Caro’s acid (peroxysulfuric acid). The mechanism is described in all text books of organic chemistry as involving an alkyl migration. Here I take a look at the scheme described by Alvarez-Idaboy, Reyes and Mora-Diez[1], and which may well not yet have made it to all the text books!
The text-book mechanism involves pathway (a, R=CF3) via species 1 and 2. A characteristic feature of many a mechanism of this type is the need for a step often labelled just PT (proton transfer). Very often, a proton will find itself attached to the wrong atom, and before the mechanism can be completed, it must be transferred to the correct location. Confusingly, there can be many ways of doing this, differing in the timing of the proton choreography. Deciding that running order can be perplexing to new students of chemistry. Tutors often will say that since PTs are very fast, it does not matter when this step occurs, since in effect all paths will lead to the final product. But we might imagine that the energies of all the various pathways can be (in principle) obtained from quantum calculations and that one will prevail over the others.
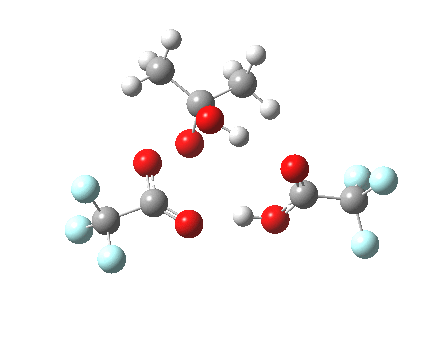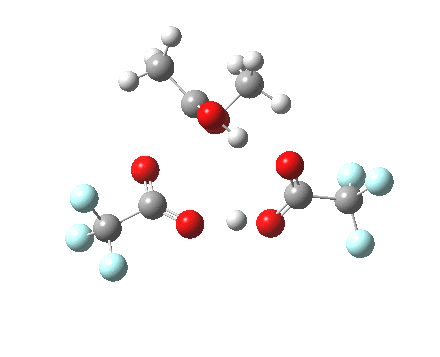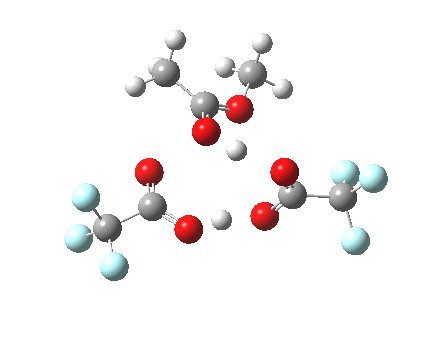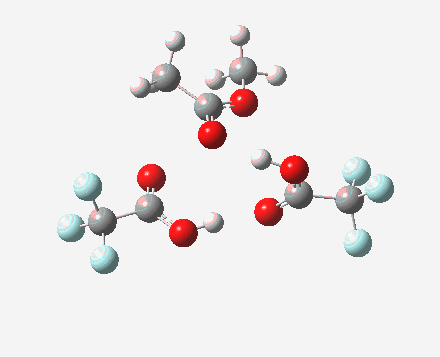
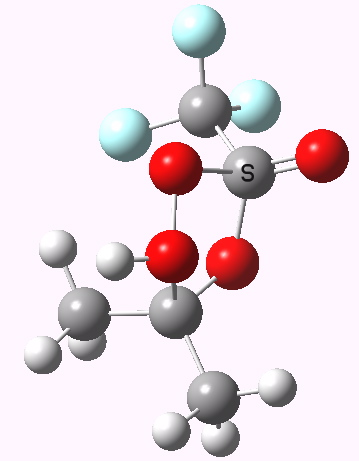
Path (b) is just one such variation, but with a twist, since it involves starting from 3 and proceeding via a cyclic transition state in which the migrating alkyl group (shown in red above) moves in concert with the relocating proton. I have repeated the original calculations (from 2007) using a somewhat updated procedure, much in the same way that the transition state for the aldol reaction was. A ωB97XD/6-311G(d,p)/SCRF=dichloromethane calculation[2] of step (b) gives the transition state and associated intrinsic reaction coordinate (IRC) shown below.
|
Cyclic 7-ring mechanism for the Baeyer-Villiger. Click for 3D. |
|
Choreographically, this transition state is quite complex. Five bonds, all different in some aspect, are changing in asynchronous concert.
Path (c) is another variation, where an extra molecule of acid (X1, 4) helps catalyse the reaction, this time by creating an 11-membered ring 4 leading to a transition state with potentially two proton transfers as well as the alkyl migration.[4] By involving an additional second molecule of acid as catalyst, we now have seven participating bond changes. Whilst the original path (b) six endo and two exo electrons move in a cycle which is tantalisingly close to but not quite pericyclic (?), path (c) extends this by four electrons. It might be tempting to try to apply a selection rule here (such as 4n+2) but I am not sure it would be justified.
|
Baeyer-Villiger, 11-ring transition state. Click for 3D |
|
To directly compare the energies of paths (b) and (c), we can repeat (b) with the addition of a more passive acid catalyst, in four new positions 3, X2 – X5. None of these are lower than 4 itself. There is one more surprise. Species 1 is not actually a minimum, but rearranges to e.g. the cyclic ring shown below. Its free energy is still higher than that of 3.
I will end with the following speculation. The point of interest to most students of the Baeyer-Villiger reaction is not the nature of the actual transition state, but deciding which of the two possible alkyl groups will migrate (in the example above both are methyls, but if one were e.g. phenyl it would migrate in preference to the methyl). The transition state teaches us that the group antiperiplanar to the O-O bond migrates. Can a system be devised where the antiperiplanar preference takes precedence over the migratory aptitude? For example, based on the following[5] (click to see 3D structure below in which one R group is clearly pre-disposed to migrate in preference to the other).
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
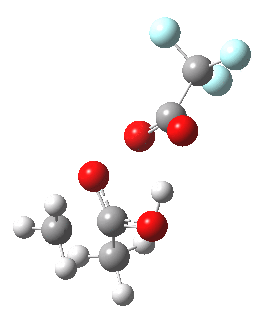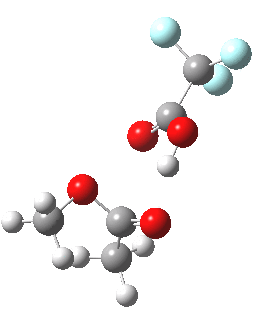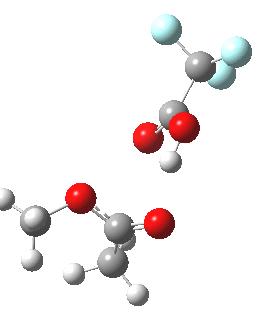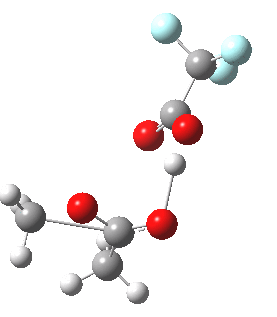{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
View Comments
Professor Henry Rezpa,
Just a couple of very naïve questions:
Why would phenyl migrate in preference to the methyl? And the hydrogen in relation to these two?
And can the molecular weight of a substituent influence the rate at which it migrates, if we could disregard the stabilization or not of the intermediary state due to its electronic properties? The logic (or not) is that hydrogen would migrate faster…
Thank you very much.
The accepted wisdom for the greater migratory aptitude of phenyl is that the transition state can adopt a spiro-coordination where delocalisation into the pentadienyl part of the phenyl brings additional stabilisation.
As for H, being light as you say, it can also tunnel in a manner that heavier groups cannot. Classical transition state theory does not include the weight of the groups involved, excepting that this will impact upon their entropy. But it is increasingly recognised that the molecular dynamics of the system has a significant role to play, and more and more examples are coming to light where such dynamics does indeed control the outcome.