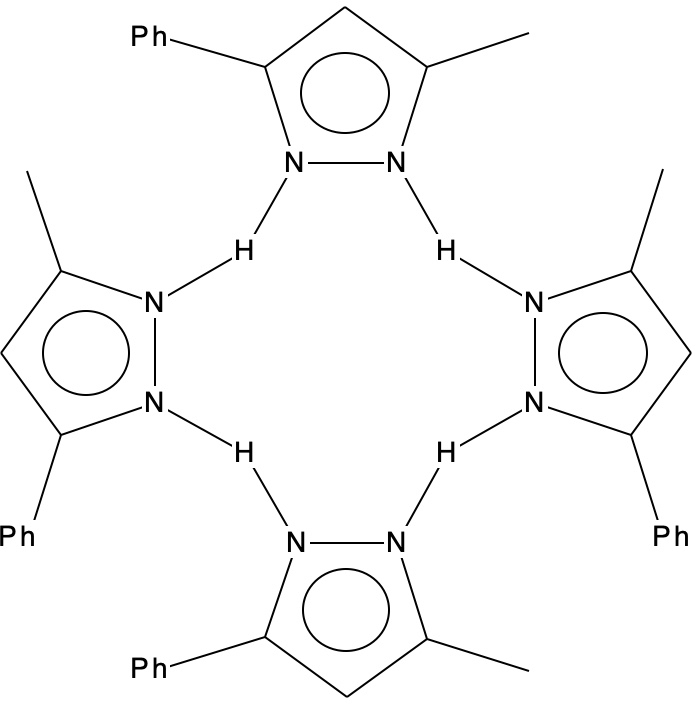
Previously, I explored the unusual structure of a molecule with a hydrogen bonded interaction between a phenol and a pyridine. The crystal structure name was RAKQOJ and it had been reported as having almost symmetrical N…H…O hydrogen bonds. This feature had been determined using neutron diffraction crystallography, which is thought very reliable at determining proton positions. Another compound with these characteristics is 3-methyl-5-phenylpyrazole or MEPHPY01.[1] Here the neutron study showed it to apparently have the structure represented below, where the solid N-H lines indicate a proton equidistant between two nitrogens.
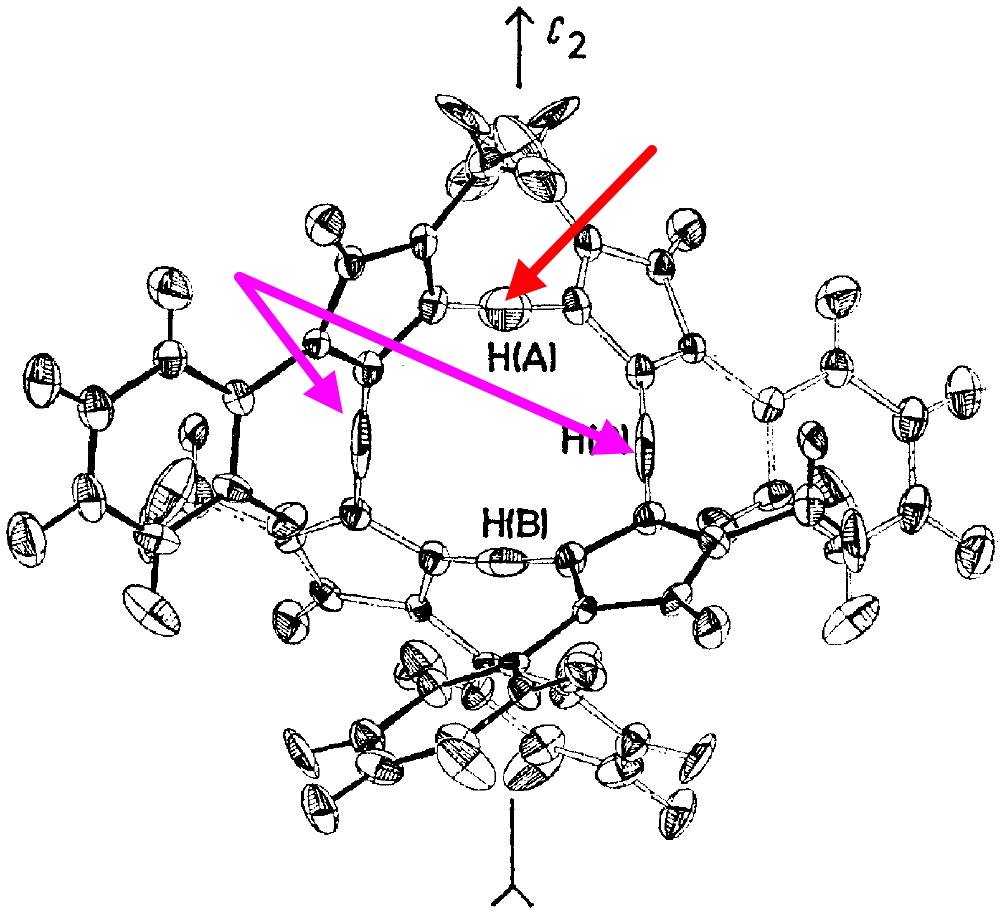
Inspection of the ORTEP plot shows a very odd feature; the thermal elipsoids (red arrow) for two of the N-H-N motifs are more or less spherical, indicating little thermal motion (the temperature of the determination is not noted, and is assumed as probably room temperature) but the other two (magenta arrows) are highly elongated in the direction of motion between the two nitrogen atoms. This feature was largely unexplained at the time of publication (1975) and indeed to this day. Here I offer a possible insight into this enigma.
The conventional structure is shown below showing four N-H bonds and four H…N hydrogen bonds. 
So now for the results of some calculations. Computed at various B3LYP(±GD3BJ)/Def2-SVPP/Def2-TZVPP levels (Table, FAIR data DOI: 10.14469/hpc/10406), the located minimum in the total energy, saddle=0, corresponds to the conventional proton-localized structure shown above, where all four hydrogens are firmly attached to the four nitrogen atoms by a regular bond and the distances are 1.032 for the NH and 1.855Å for the hydrogen bond it forms. A zwitterionic isomer comprises the ion-pair shown below, examples of each component of which are known in the CSD (crystal structure database).

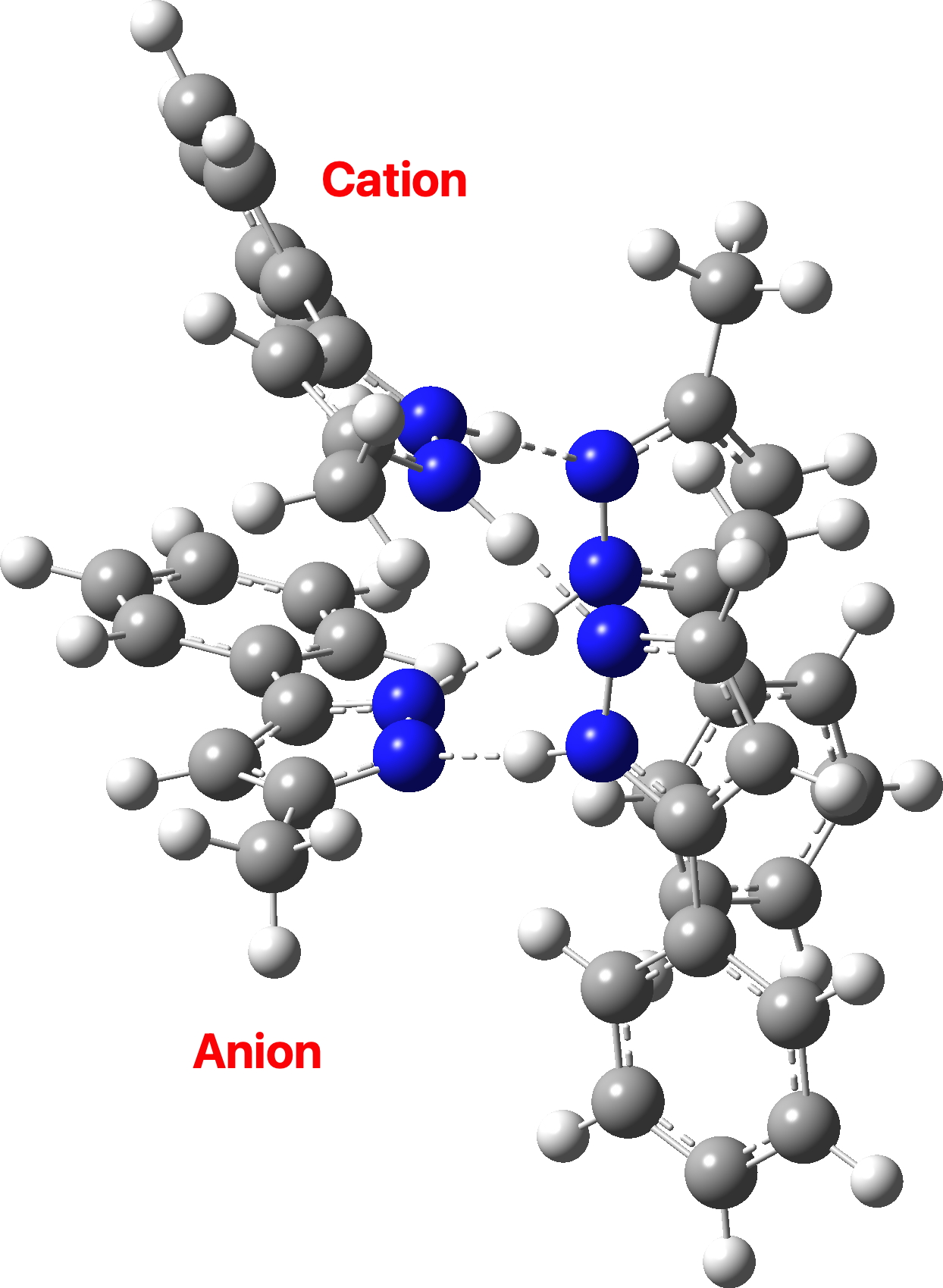
There are three ways of distributing this motif, of which only 1 is stable to proton transfer. Structure 2 has a higher degree of charge separation whilst 3 superficially appears to reduce the degree of charge separation compared to 1. In fact, the three-dimensional structure of 1 allows the negative ion to stack above the positive ion, thus actually achieving minimal separation of charges.

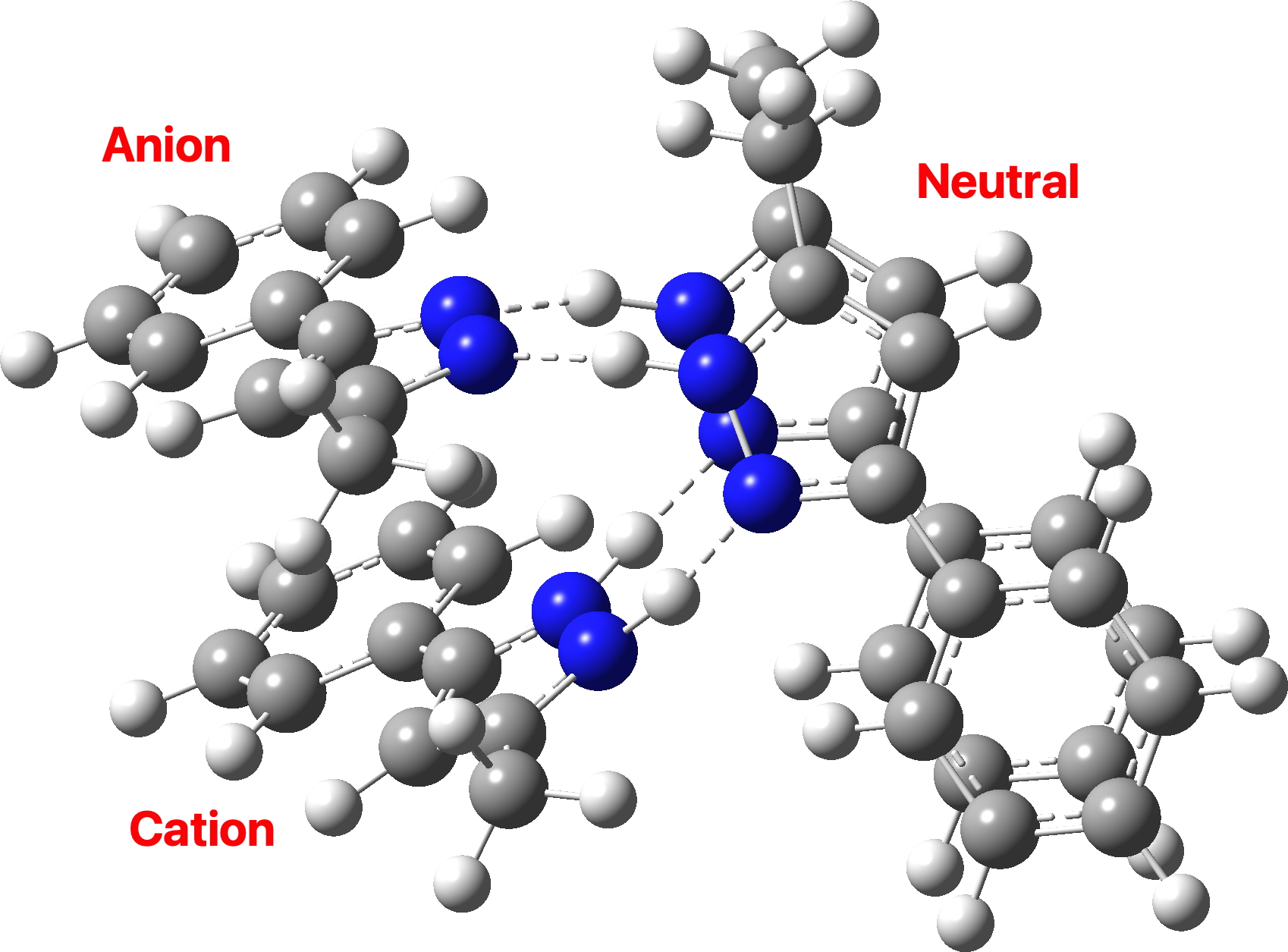
The stacking also depends on the type of calculation. If dispersion correction is included, the aromatic faces stack directly above each other (as above). If omitted, the stacking actually corresponds more closely to that observed in the reported crystal structure, since the attraction between faces occurs not only within a structure but between adjacent structures in the solid state (something not modelled when the dispersion correction is applied only to a single unit).
The lesson learnt from the previous post is that the position of protons as determined by quantum-chemical geometry location using minimisation of the total computed energy might be misleading. Better perhaps to use the computed free energy? When this is done, as we saw in the previous post, the transition state for proton transfer as located in the total energy surface can actually have a free energy that is lower than that of the total energy minimum. So, for MEPHPY01, a stationary point in which all four hydrogens correspond to the apparently symmetrical experimental neutron diffraction structure emerges as saddle=3, corresponding to three force constants being negative. The bond lengths for this geometry occur in pairs, two with NH 1.25/N…H 1.30 and two with 1.28/1.28, revealing interesting asymmetry.
The normal vibrational modes for these three -ve force constants are shown below. The first (ν 1315i cm-1) shows all four hydrogens exchanging between nitrogens, a quadruple proton transfer. The second (ν 896i cm-1) shows a double proton transfer between one pair exchanging between two nitrogens and the last (ν 801i cm-1) is similar in form, but shows the other pair exchanging between a second different pair of nitrogens. These last two vibrational modes correspond to the very thermal ellipsoids seen in the crystal structure diagram at the top, where one pair of hydrogens show little motion and the other pair involves much greater motion between a pair of nitrogen atoms.This would correspond to formation of a species exhibiting two conventional NH…H hydrogen bonds and two symmetrical N…H…N units.
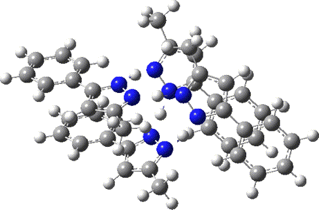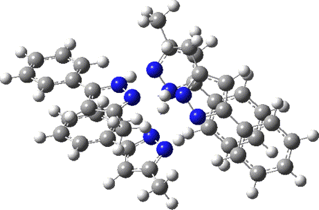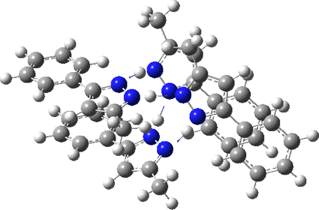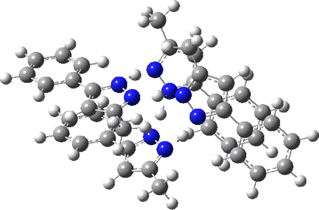
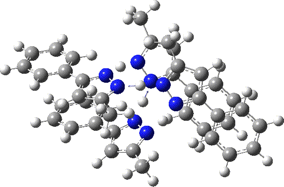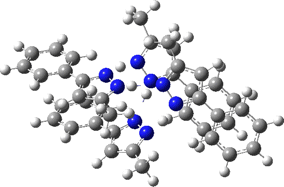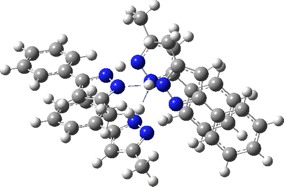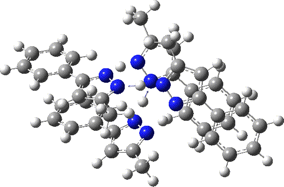
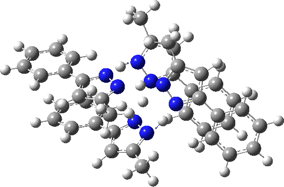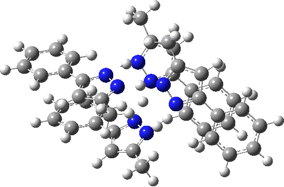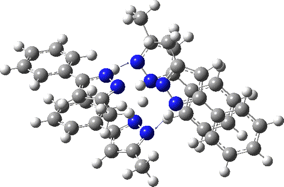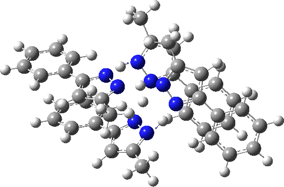
Two further stationary points corresponding to saddle=2 and saddle=1 can also be located (Table).
| stationary points | B3LYP+GD3BJ/gas | B3LYP+GD3BJ/DCM | B3LYP/gas |
|---|---|---|---|
| SVPP, saddle=0, neutral | -1984.448365(0.0) | -1984.460070(0.0) | -1984.241838(0.0) |
| SVPP, saddle=0, ion-pair | -1984.426827(13.5) | -1984.439305(13.0) | -1984.218379(14.7) |
| TZVPP, saddle=0, neutral | -1986.712743(0.0) | -1986.725277 (0.0) | -1986.509333(0.0) |
| TZVPP, saddle=0, ion-pair | -1986.688467(15.2) | -1986.703562 (13.6) | -1986.481384(17.5) |
|
|
|||
| SVPP, saddle=1 | -1984.428898(12.1) | -1984.442004(11.3) | -1984.220238(13.6) |
| SVPP, saddle=2 | -1984.430085(11.5) | -1984.442986(10.7) | -1984.220968(13.1) |
| SVPP, saddle=3 | -1984.431457(10.6) 1315, 896, 801 |
-1984.441075(11.9) 970, 603, 294 |
-1984.223176 (11.7) 1335, 782, 760 |
| TZVPP, saddle=1 | -1986.691518(13.2) | – | |
| TZVPP, saddle=2 | -1986.689979(14.3) | – | |
| TZVPP, saddle=3 |
-1986.690265(14.1)
|
– | |
Now here is the wacky thing. At the gas phase SVPP basis set ± dispersion levels, these lower-order saddle points are actually HIGHER in free energy than the third order saddle point! Conventional wisdom is that the higher the order of the saddle point, the higher should its energy be! I am not aware of anyone reporting an inverse observation before. The effect however is solvation and also basis-set dependent, since adding dichloromethane as a continuum solvent changes the free energy minimum from the third to the second-order saddle point. It might well also be dependent on the density functional method.
What are we to conclude? The free energy barriers for all the proton transfer saddle points computed above are not that small, being ≥ 10 kcal/mol. But at room temperatures, these exchanges will in fact be fast kinetic processes and the measured neutral diffraction structure may well emerge as averaged in some way. The free energies of the higher order saddle points suggests the dynamics of this system may in fact be very complex and very different from any “normal” hydrogen bonded system. This is clearly not the final word yet, but it does hint that the proton transfer dynamics of 3-Methyl-5-phenylpyrazole may be a system very well worth looking at again! And indeed exploring how robust the effects noted above are to different density functionals.
This post has DOI: 10.14469/hpc/10512
Author
References
- F.H. Moore, A.H. White, and A.C. Willis, "3-Methyl-5-phenylpyrazole: a neutron diffraction study", Journal of the Chemical Society, Perkin Transactions 2, pp. 1068, 1975. https://doi.org/10.1039/p29750001068