The Bürgi–Dunitz angle is one of those memes that most students of organic chemistry remember. It hypothesizes the geometry of attack of a nucleophile on a trigonal unsaturated (sp2) carbon in a molecule such as ketone, aldehyde, ester, and amide carbonyl. Its value obviously depends on the exact system, but is generally taken to be in the range 105-107°. A very good test of this approach is to search the crystal structure database (this was how it was originally established[1]).
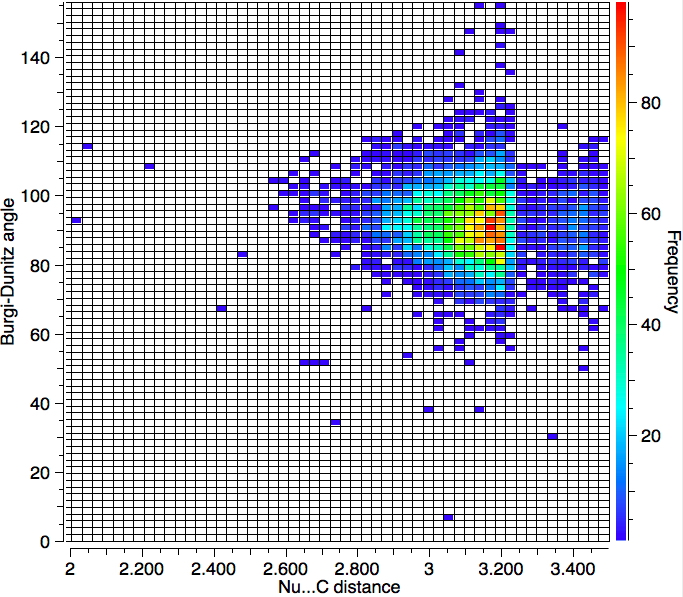
With no temperature specified, 6994 hits are obtained as below. So the most probable angle (red spot) is ~90°.
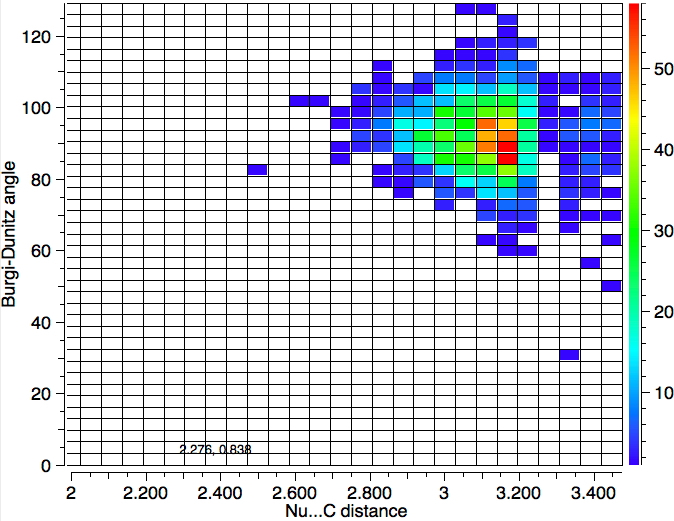
One important change to the search is to decrease the temperature to 120K, since structures will have less vibrational noise. The number of hits decreases to 1279, but the most probable angle if anything reduces slightly.
So we have something of a mystery; this crystallographic data shows an angle of approach about 15° less than the oft quoted value. Here are some thoughts:
Well, to get to the bottom of this will require reducing the scope of both QA and QB, to find which if any of discrete values for these two variables can indeed give an angle of 105-107°. This would make for quite a good student group project; I expect a group of 8 students could sort this out quite quickly!
This post has been cross-posted in PDF format at Authorea.
In an earlier post, I discussed a phenomenon known as the "anomeric effect" exhibited by…
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
{kind=link}
{kind=link}
View Comments
Could you tell me, what structural data base did you use during your searches?
The Cambridge Crystal Structure Database.
This is a really nice exercise for students (or any reader in general) to revisit the published data and theory critically!
I believe that the primary source of the discrepancy comes from the overwhelming number of structures featuring antiparallel C=O interactions. In those cases, the O of one moiety sits on the C of the other, resulting in O...C=O angle at ~90 degrees.
Restricting the nucleophile to tertiary amines will significantly clean up the search results (but reducing the number of hits). Also, CSD Conquest searches by default only consider intermolecular contacts; this setting can be changed to include the intramolecular cases. This search is closer to the original Bürgi–Dunitz papers (JACS 1973, 95, 5056 and Tetrahedron 1974, 30, 1563, where 6 structures were examined!). A Conquest query file and the hitlist of such a search can be downloaded here: https://bit.ly/2SVxjWX ; the mean value of this angle is about 104 degrees.
The possibility of hydrogen bonding in the case of primary and secondary amines and the various O nucleophiles will undoubtedly complicate the analysis. The latter has been partially examined in Bürgi–Dunitz's paper in Acta Cryst. 1974, B30, 1517.