This is a continuation of the discussion started on Steve Bachrach’s blog about a molecule with a very short H…H interaction involving two Si-H groups with enforced proximity. It had been inferred from the X-ray structure[1] that the H…H distance was in the region of 1.50Å. It’s that cis-butene all over again! So is that H…H region a bond? Is it attractive or repulsive? Go read Steve’s blog first.
Next, in the previous post, I had blogged about assigning a publication doi to a procedure or tool. So Steve’s post provided a good opportunity to show how this might work. This is the tool doi: 10.6084/m9.figshare.811862 Using it, and another doi, this time data: 10.6084/m9.figshare.812621 we can create a new data set, visualised below.‡ This is the NCI (non-covalent-interaction)[2] isosurface of the reduced density gradient, and colour coded according to (λ2)ρ, the eigenvalue of the density Hessian, to indicate attraction or repulsion. You should know that according to this scheme, blue is strongly attractive (it is normally seen for example for strong hydrogen bonds). You can see the blue region in-between the H…H region. So a strong (di)hydrogen bond then!
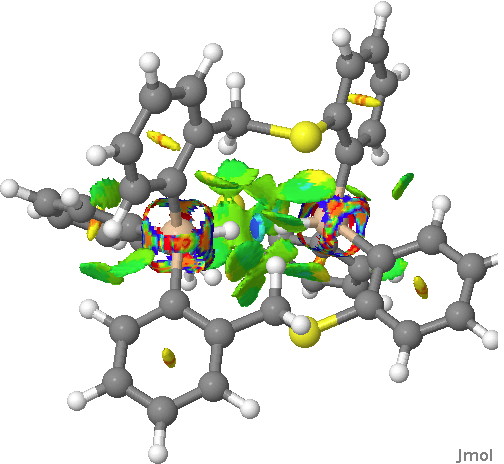
Click for 3D.
Well, interesting, and this needs to be looked into further. For example, it might in fact be an anomalous result since the H…H region may in fact have charge-shift character,[3] which can change the characteristics of the density Hessian (and its Laplacian).
One more property, the NBO (natural bond orbitals) for this region. Can one tell if H…H is bonding? It might seem so. Finally, the Wiberg bond index for the H…H region is 0.027, very slightly bonding.
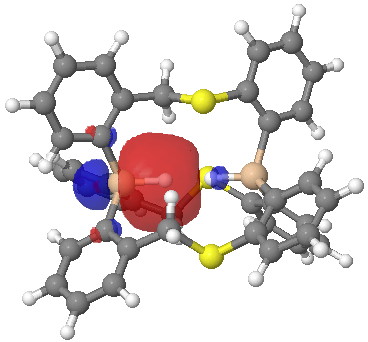
Click for 3D.
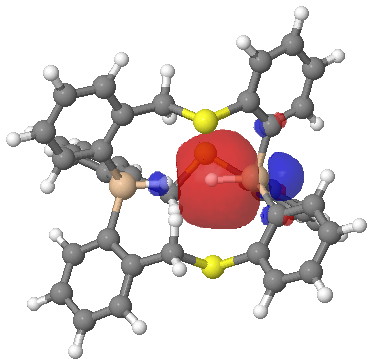
Click for 3D.
I should conclude by stating that whilst the initial discussion of this molecule took the form of comments on Steve’s blog, the nature of the Word press system used there (and here) does not allow commentators to insert rotatable models into comments. So that discussion is continued here in order to achieve that effect.‡
‡ And yet another data-doi could be created showing the interactive display, and this could be transcluded back into Steve’s blog to continue the to and fro.
Postscript: I have added the QTAIM analysis that first appeared on Steve’s blog. The red arrow points to the H…H bcp. The blue arrow points to one of the other (three in all) bcps, all of which are very close to a ring critical point, and hence should be regarded as unstable and prone to annihilation.
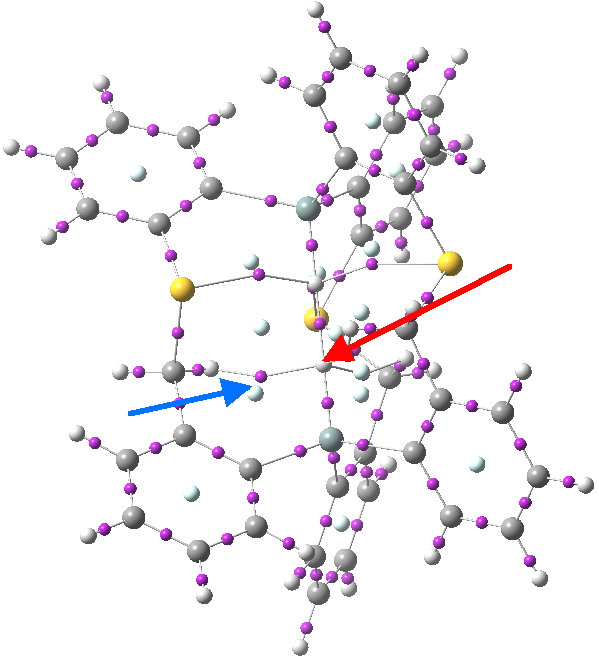
Click for 3D.
The B3LYP+D3/TZVP calculated Si-H vibrations are shown below. The VCD spectrum[4] is shown below it.
Click for animation

Postscript 2. The Si-H vibration is reported as 2325 cm-1 (with weak bands at ~ 2360-2380), but in footnote 7 of the original report[1] the authors do note that there should be two Si-H bands. The calculation shown above suggests two values, 2406 (intensity 37) and 2460, intensity 10).
The 1H NMR spectrum of the two Si-H bands has two singlets which are reported (but not discussed in the text anywhere) as 8.23 and 8.56 ppm (Δδ 0.33ppm, CDCl3); B3LYP+D3/TZVP calculation[5] predicts 8.79 and 9.40 (Δδ 0.61 ppm). It is possible however these shifts are perturbed by spin-orbit coupling from the silicon[6]. The diastereotopic methylene groups are reported as 4.10 and 4.83, calc 3.95 and 5.03 ppm, which is a reasonable match, and gives confidence to the theoretical prediction.
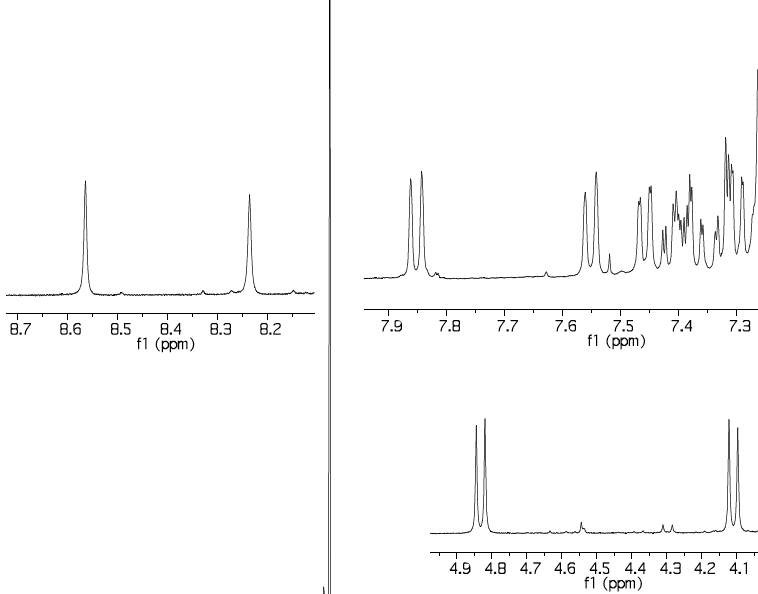
The 29Si NMR is reported as -32 and -40 ppm, calculated -31.6 (for the Si-H associated with bridging S) and -39.9 (for the Si associated with bridging C) ppm, which matches very well.
The reported 1H NMR spectrum appears to show 29Si satellites but their values are not reported numerically in the article. In Ph3Si-H, this coupling is known to be ~±205 Hz. The 29Si-1H couplings are calculated for the compound above as -191 (for the -31.6 peak) and -166 Hz (for the -39.9 peak), this latter being notably lower than the former. The 29Si-29Si coupling is 10 Hz. Most interestingly, the 0JHH coupling (i.e. through space) is predicted as +5.4 Hz; there is no sign of such a coupling for the two singlets reported at 8.23 and 8.56 ppm. This last observation may be of significance in terms of whether the axis along the four atoms Si-H-H-Si is indeed linear, or whether it is bent (enabling the two hydrogens to avoid close contact).
This problem is not yet closed!
Author
References
- J. Zong, J.T. Mague, and R.A. Pascal, "Exceptional Steric Congestion in an <i>in</i>,<i>in</i>-Bis(hydrosilane)", Journal of the American Chemical Society, vol. 135, pp. 13235-13237, 2013. https://doi.org/10.1021/ja407398w
- E.R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A.J. Cohen, and W. Yang, "Revealing Noncovalent Interactions", Journal of the American Chemical Society, vol. 132, pp. 6498-6506, 2010. https://doi.org/10.1021/ja100936w
- S. Shaik, D. Danovich, B. Silvi, D.L. Lauvergnat, and P.C. Hiberty, "Charge‐Shift Bonding—A Class of Electron‐Pair Bonds That Emerges from Valence Bond Theory and Is Supported by the Electron Localization Function Approach", Chemistry – A European Journal, vol. 11, pp. 6358-6371, 2005. https://doi.org/10.1002/chem.200500265
- H.S. Rzepa, "Gaussian Job Archive for C39H32S3Si2", 2013. https://doi.org/10.6084/m9.figshare.818954
- H.S. Rzepa, "Gaussian Job Archive for C39H32S3Si2", 2013. https://doi.org/10.6084/m9.figshare.817910
- D.C. Braddock, and H.S. Rzepa, "Structural Reassignment of Obtusallenes V, VI, and VII by GIAO-Based Density Functional Prediction", Journal of Natural Products, vol. 71, pp. 728-730, 2008. https://doi.org/10.1021/np0705918
Tags: Postscript, Steve Bachrach, X-ray
The spin-spin coupling constants computed and reported in the post above were done at the B3LYP/TZVP level. They are however known to be sensitive to basis set. A procedure that decontracts the basis to correct for this effect can be applied (doi: 10.1021/ct600110u, implemented in Gaussian as NMR(spinspin,mixed) ). This results in the following predicted couplings;
29Si-H, -245 and -210 Hz; 29Si-29Si, +11.1, 1H-1H +6.2 Hz.
This calculation can be found at doi: n8r.
Bob Pascal has kindly sent me the measured J(SiH) couplings, as ±233 (δ8.57) and ±206 Hz (δ 8.24). These compare with calculated values of -245 and -210 Hz respectively.
Bob has sent me some more data, this time on an analogue (below) where a P replaces one of the S-H groups, and suggested I post them here to continue the discussions. These results come from an article about to be published in Tetrahedron (doi: tba).
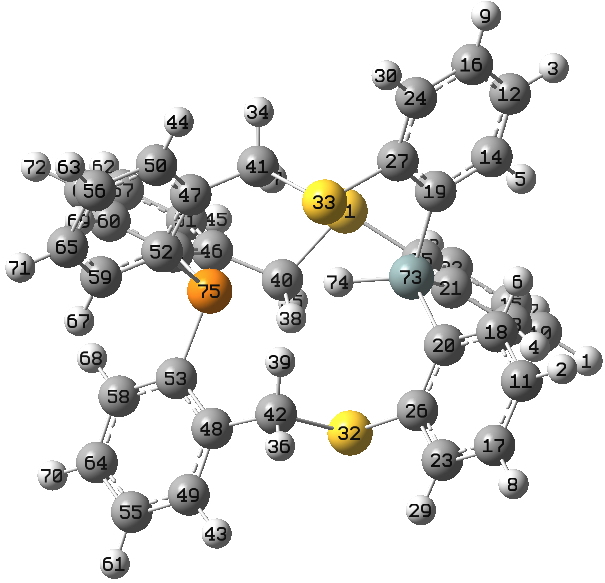
In order to further calibrate the NMR predictions, this system has the following measured values: δ29Si -35.8, 31P -48.9, and 1H 9.31 ppm. J(SiH) = ±248 Hz, J(SiP) = ±76 Hz, and J(PH) = ±25 Hz
The calculated values are: δ29Si -34.9, 31P -42.7, and 1H 10.1 ppm. J(SiH) = -262 Hz, J(SiP) = -87 Hz, and J(PH) = -29 Hz.
The most interesting of these is the through-space PH coupling. In truth, I have no idea if this is unusually large or not; does anyone know?
Computation allows one to ask what if. So, “what if one replaced the -S- bridge in the Si-H…H-Si system with an oxygen bridge“?
The NMR reveals a through-space H-H coupling of +8.0Hz, and a H-H distance of 1.47Å, which would be another record. Whether this system can be made synthetically is another question however.
The IR has two Si-H stretches of 2452 and 2523 cm-1 which also reveals a stable minimum with no distortive imaginary modes.
Roald Hoffmann has pointed out to me modelling of SiH4 at high pressures. It takes 150 GPa (~1.5 million atmospheres) to shrink the H…H distance to 1.5Å or less. Since this around the value reported by Pascal et al, this implies the internal compression is equivalent to ~1.5 million atmospheres. Pretty impressive!
“Any ideas for a compound with an even shorter non-bonded H…H distance?”
Yes, there is! See my joint paper with Dr. Firouzi just posted to arxiv: http://arxiv.org/abs/1310.5375 for examples of shorter non-bonded H…H distances. we have reach to distances as short as 1.38 Å however, I think even shorter distances are also possible to be realized at least computationally.
In response to the previous comment, I show a diagram of one of the species suggested there. Whether this system could be synthesized is an interesting question; it does look very highly strained. I would also observe that the C-H-H-C alignment below is very non-linear, whereas the Si-H-H-Si is completely linear.
I also calculated the H…H through space NMR coupling, and this comes out as +4.6 Hz, lower than the Si-H…H-Si coupling of ~6Hz (but is this because it is non-linear?). This might also suggest that the predicted magnitude of the Si-H…H-Si coupling is associated in part at least with the nature of the Si atom (and any spin-orbit effects that might accrue for this element, but not for C). Clearly, a high level calculation, which includes such corrections, does seem a desirable objective to clarify this point.
“Whether this system could be synthesized is an interesting question; it does look very highly strained.”
Indeed. I think the strain energy (MM methods) or alternatively the energy of formation (QM methods) must be calculated to see whether there is any chance for this and other proposed systems to be synthesized although the “kinetic stability” is also an important issue. These are rigid hydrocarbons so there is no problem with conformational variations however, the case of severe deformation can not be excluded and this may ruin the kinetic stability. For the case of B6C(-2) some years ago I have done such project (J. Phys. Chem. A, 2008, 112 (41), pp 10365–10377) (check the DOI: 10.1021/jp806874u) and considered all deformation pathways so I know it is simple to say but hard to do such calculations! To realize linear CH…HC form, I think “3D” hydrocarbon skeletons must be designed (probably Pascal’s own design is somehow an example for using 3D structures) to put “pressure” from below and above on CH…HC making it impossible for severe non-linear deformations toward a less crowded corner.
It is strange that these and similar problems have not yet been considered computationally…
The molecule above might be susceptible to homolytic cleavage, and I note that one can try to estimate the energies for this sort of process according to the procedures outlined at 10.1021/jz401578h for in effect the dissociation of F3SSF, by systematically testing each bond.
thoughtful idea…
Regarding the role of linear vs. non-linear CH…HC arrangements, though not for ultrashort H…H contacts, there are useful discussions at 10.1021/ct400070j and 10.1038/nchem.1004
Regarding the “3D” structures that may be used to force certain arrangements of atoms I found these two works from Leo Radom in my files on exotic hydrocarbons: Angew. chem. Int. Ed. 38, 2876 (1999) and Pure. Appl. chem. 70, 1977 (1998). Although they are not directly relevant to above discussions but demonstrate how design in “3D” may be used to achieve a certain configuration of atoms.
Going back to the discussion of the original Si-H..H-Si close contact, a calculation for a slipped (higher energy) isomer reveals that the 0JHH coupling is -3.6 Hz (H…H distance 1.78Å). Recollect that for the linear form, the predicted coupling was +6.2Hz, and therefore that slippage tends to reduce the coupling. Presumably at some level of such slippage, it reduces to almost zero. I mention this because the original experimental spectrum showed no discernible coupling. Perhaps a small degree of slippage, perhaps due to thermal libration or other vibrational effect, might be responsible. I offer it merely as one suggestion.
Response posted on behalf of Bob Pascal. Part 1.
———————————–
Some time ago, Dr. Rzepa drew my attention to the discussion of our in,in-bis(hydrosilane) (1; Zong et al., JACS 2013, 135, 13235) on a couple of blogs, including his own. Of particular interest to me was the discussion of possible spin-spin coupling between the axial hydrogen atoms of compound 1, which was certainly not apparent in the experimental 1H NMR spectrum of this material.
We have previously observed strong “through-space” scalar coupling of a variety of atoms in several of our in-cyclophanes, and Frank Mallory observed many more examples long before we ever made a cyclophane. However, in every example that I can recall, at least one of the coupled atoms possessed lone pair electrons. We have made many crowded molecules, but never have we seen anything that resembled an H…H “through-space” coupling.
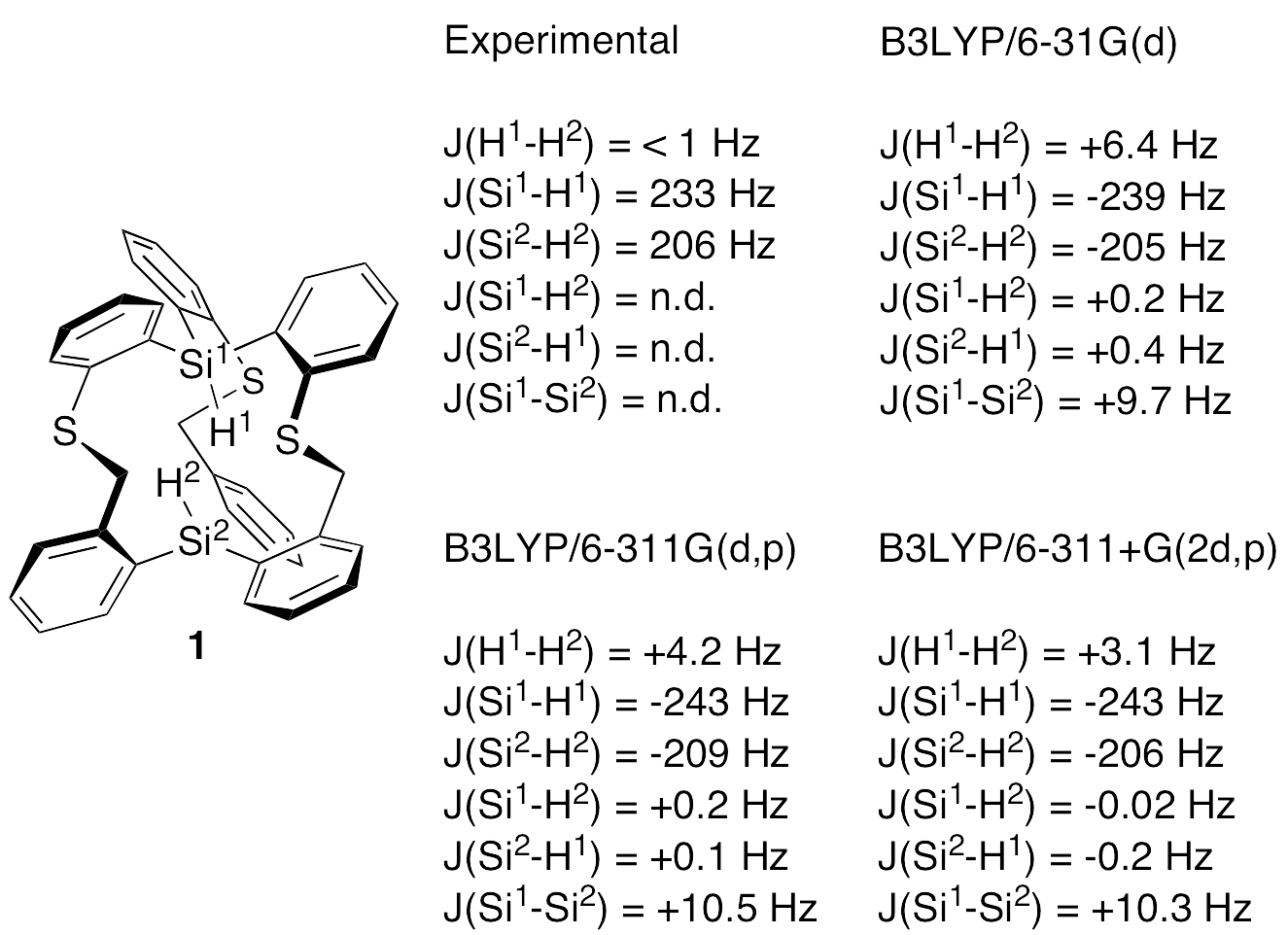
Experimental and calculated coupling constants for the axial atoms of compound 1 are listed below. Simple HF and HDFT methods give reasonable values for the Si-H one-bond coupling constants, and Dr. Rzepa has obtained even better agreement with experiment by using B3LYP/TZVP (see his blog). However, H…H coupling was not observed experimentally, at least not of the calculated magnitude.
We have only a few milligrams of compound 1, and most of it is tied up in crystallization experiments (for neutron diffraction). However, my student Jie Zong had just prepared a 1 mg sample of 1 when we received Dr. Rzepa’s message, and we used it to take a hard look at possible axial H…H coupling. The 1H NMR spectrum of 1 reproduced in the Supporting Information of our paper did not reflect especially good shimming, as the lack of meta-couplings in the aromatic proton resonances of 1 makes obvious. Of course, this would not have obscured a 7 Hz coupling constant (Dr. Rzepa calculated JHH’s of 5.4 Hz and 6.2 Hz with B3LYP/TZVP using two slightly different methods), but a spectrum recorded under conditions of higher resolution was needed to define better this interaction.
Figure 1. Expansion and decoupling of the Si-H resonances in the 300 MHz 1H NMR spectrum of 1.
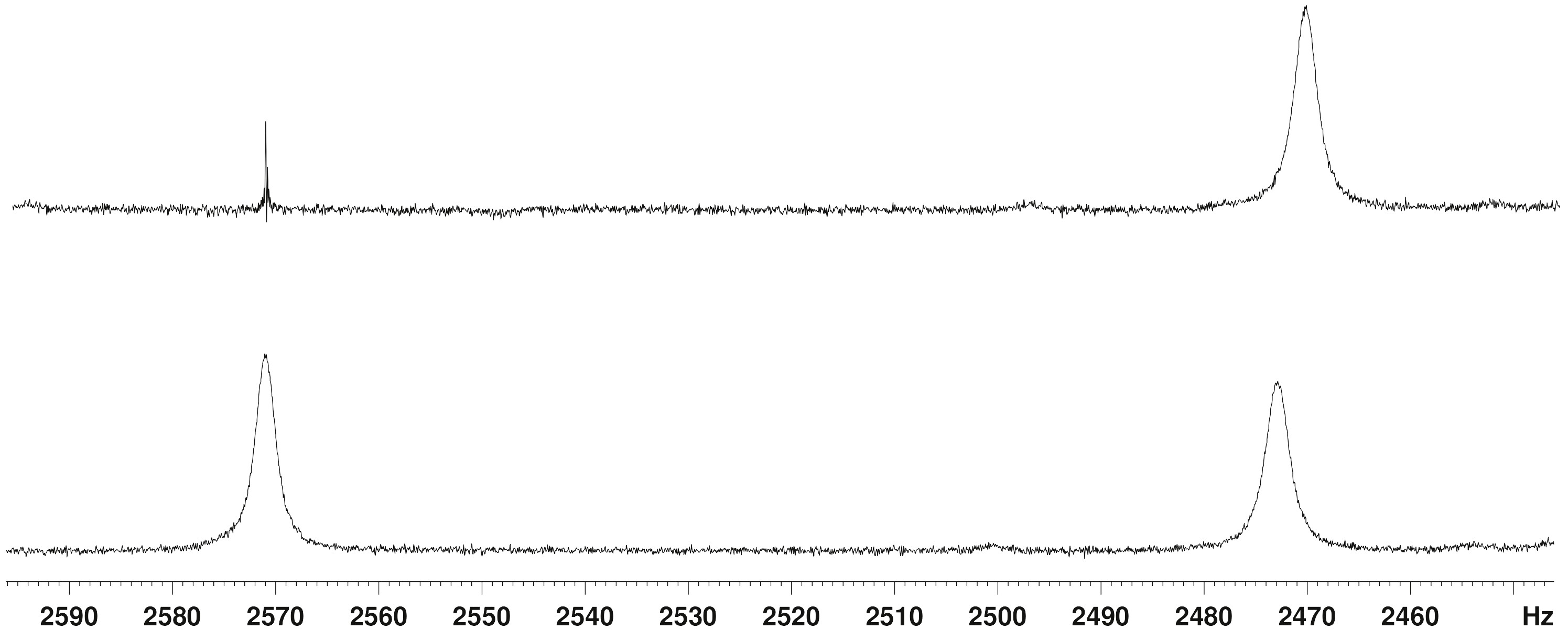
Figure 1 shows an expansion of the Si-H region of a 1H NMR spectrum of 1 in which the residual CHCl3 resonance has a line width of 0.4 Hz and the aromatic meta-couplings (ca. 1.5 Hz) are clearly seen (neither shown in this figure). In the lower spectrum, the line widths of the δ 8.24 and δ 8.57 resonances are 2.7 Hz and 2.4 Hz, respectively, with no sign of any fine structure. The upper spectrum is a spin decoupling experiment, but irradiation of the δ 8.57 resonance produced no change in the line width of the δ 8.24 resonance (although there was a significant NOE enhancement). A JHH of 7 Hz is therefore out of the question, and even a 1 Hz upper limit on any H…H coupling constant seems generous. The observed broadening of the Si-H peaks is easily accounted for by conventional, long-range, through-bond coupling to the aromatic and methylene proton resonances, the largest of which is calculated to be 0.5 Hz. We also recorded a COSY spectrum of this sample, and it showed very weak cross peaks corresponding to Si-H…H-Si coupling. A few similarly weak cross peaks were observed between the Si-H peaks and other resonances in the spectrum, and thus we estimate JHH to be ca. 0.5 Hz at present, but this may change when we have additional compound 1 and are able to record a better COSY spectrum.
Possible explanations of the lack of observed coupling (suggested by Dr. Rzepa, who can speak more fully about them) include (1) an as yet undetected, low-energy conformation of 1 with no close H…H contact, either as the true global minimum or in rapid conformational exchange with the observed C3 structure, (2) sufficient off-axis libration of the hydrogen atoms to widen their average separation. I am partial to a third explanation: inadequate computational methodology.
What about other cyclophanes? We attempted to detect H…H couplings in several of our other in-cyclophanes, but none were observed. However, the largest of these coupling constants was calculated to be only 0.9 Hz; thus the failure is not so surprising.
But what is the evidence in the literature for strong H…H “through-space” coupling? I would have been a lot less concerned about the discrepancy between the experiment and calculations for compound 1 had I only known to look at Figure 1 in Anet and Dekmezian, JACS 1979, 101, 5449:
Spectrum A is a comparison of the line widths of Ha in a mixture of compounds II and II-d, which are derivatives of the pentacyclododecane that previously held the world record for closest non-bonded H…H contact. We know from neutron diffraction that Ha and Hb are only 1.62 Å apart. In the 348 MHz 1H NMR spectrum shown, Ha in II is a singlet with a line width of only 1.0 Hz, and deuteriation of Hb (II-d) reduces the line width to perhaps 0.6 Hz. This wonderful example of “chemical spin-spin decoupling” places the “through-space” coupling constant J(Ha-Hb) at about 0.5 Hz. (The difference in line width is 0.4 Hz, but about 0.1 Hz should be added due to deuterium coupling to Ha.) If so crowded a molecule shows only this tiny coupling constant, then it’s no wonder that H…H “through-space” couplings are hard to find!
Well, one of the chief reasons to make theoretically interesting molecules is to provide points of calibration for computational methods. Now that we have an experimentally measured “through-space” H…H coupling constant (although these atoms are separated by only five bonds, so that the small observed interaction could be purely “through-bond”), what do quantum calculations say about this interaction?
In this case, there is an obvious decrease in the calculated “through-space” coupling constant with increasing basis set size, and medium-sized, triple zeta basis sets are required to give results consistent with the experimental data. (Note: except as indicated, all geometries were optimized at the same level as the NMR calculations, and the keyword NMR=SPINSPIN was used for all calculations in Gaussian09.)
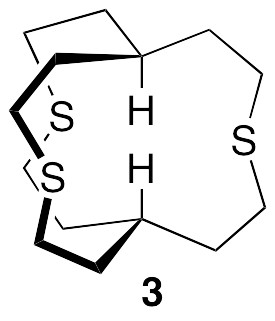
In order to explore further the basis set dependence, consider the as yet unknown model compound 3, which has a linear arrangement of axial atoms, as seen in 1, but a smaller size and higher symmetry (C3h). A similar trend of reduction of coupling constants with increasing basis set size is observed.
Posted on behalf of Bob Pascal. Part 2.
———————————-
The calculated JHH’s for both II and 3 drop sharply upon switching from double zeta to triple zeta basis sets. Thus, even though 6-31++G(d,p) is as large as 6-311G(d,p) (and aug-cc-pVDZ is much larger), the latter gives a much better estimate of the coupling constant in II (and presumably also in 3). It also seems from both examples that TZVP is perhaps not the best choice among triple zeta basis sets for coupling constant calculations. I would really like to see a B3LYP/aug-cc-pVQZ NMR calculation for compound 3, because that basis set would restore diffuse functions as well as include polarization on the core electrons. This is by no means an impossible calculation, but, since the calculation would employ 2568 basis functions, it would probably require three weeks of my computer’s undivided attention!
While crunching through these calculations, I discovered a recent paper that explicitly considers H…H “through-space” scalar couplings, all very small (< 0.6 Hz), in a variety of molecules: Dracinsky et al., “Computational and Experimental Evidence of Through-Space NMR Spectroscopic J Coupling of Hydrogen Atoms,” Chem. Eur. J. 2012, 18, 981-986. Most significantly (to me), they concluded (a) that the B3LYP/6-311++G(2df,2pd) basis set was required in order to have the calculated coupling constants match the experimental J’s, and (b) that this basis set gave results as good as the even larger basis set aug-cc-pVTZ. The recommended basis set does indeed give the lowest value of JHH for 3 (along with the ridiculously large cc-pVQZ). What is the calculated JHH in 1 if a variety of triple zeta basis sets are used for the calculation? We first carried out NMR calculations for 1 at the B3LYP/6-311G(d,p) and B3LYP/6-311+G(2d,p) levels, which seem to give good results for II and 3. The resulting axial atom coupling constants are given below. The calculated H…H coupling constants drop to 3.6 Hz and 2.5 Hz, respectively. The latter is still significant, but at least it is no longer greater than the line widths of the singlets observed in the experimental spectrum!
At this point we bit the bullet and carried out an NMR calculation for 1 at the B3LYP/6-311++G(2df,2pd) level. As tabulated above, the calculated H…H coupling constant dropped a bit further to 2.3 Hz. This may be near the limit for triple zeta basis sets, but it is by no means clear that this represents a true convergence on the calculated coupling constant.
One may speculate that the computed “through-space” H…H couplings are a sensitive measure of basis set superposition error, but they could easily be some other kind of computational artifact. I have no understanding of the inner workings of NMR calculations, but it seems likely that the electron-nuclear interactions are at best semiempirically, and more likely empirically treated. One may hope that the transmission of the coupling is what the NMR calculations do well, but what aspect of the electronic structure calculation is most important? It is clear that bigger basis sets and diffuse functions are important for better treatment of the “through-space” interactions, but is a better description of the core electrons also required?
On the other hand, since the experimental data for 1 clearly rule out a coupling constant of even 2 Hz, and since I have yet to find a single example of even a 1 Hz experimental “through-space” H…H coupling constant, I have a hard time taking any of the present calculations too seriously. Indeed, the possibility that even the small coupling constants observed in the literature are mostly due to “through-bond” effects cannot be excluded. Until someone can show me an experimentally measured “through-space” H…H coupling constant greater than 1 Hz, preferably one in which at least eight bonds separate the protons in question and where a clear doublet (not merely line broadening) is observed, I will be inclined to regard such J’s as trivial or nonexistent and the calculations that purport to demonstrate their existence as inadequate to describe reality. Does anyone know of such an example?
As you can see from the above, Bob sent me a wonderful and generous response (which I had to split into two, because it exceeded the length of a comment allowed by WordPress!).
We have reached a delightful place (in a sense), where we need to find out if we can trust the theory (which seems to be telling us that the H…H coupling should be significant) or the experimental results, which are telling us it is small.
Of course there is another variable, which is the model used to interpret the experiment. All the above is in effect a static model, in which the alignment of the four atoms Si-H…H-Si is precisely linear. In that orientation, theory predicts a significant H…H coupling. But of course the molecule in a dynamic sense does not spend ALL of its time in this precise linear orientation. And the calculation I reported in an earlier comment seems to imply that as this orientation deviates from co-linearity not only does the coupling decrease, it actually becomes negative! Perhaps for only modest deviations from co-linearity it is actually precisely zero!
So one possible explanation is that the molecule “spends enough time” in libration-induced slightly non-linear orientations of the crucial four atoms that the time-averaged effect is to significantly reduce the coupling? I would argue that the theory gets the OTHER couplings pretty well (albeit over-estimated using the NMR(spinspin,mixed) method, which improving the basis set should in fact correct). So is there any particular reason to distrust just one of them? Perhaps there might be!
Another, even more interesting explanation is that the H…H region is highly compressed. One might assume that compression will increase electron correlation. Perhaps the standard correlation as described by B3LYP-DFT cannot cope well with this region. Unfortunately, no equivalent to the NMR(spinspin,mixed) GIAO computation for MP2 or MCSCF methods is available.
At any rate, we do now have at least two further avenues to explore theoretically. I am perhaps not quite so pessimistic that the theory is entirely to blame for the mis-match between predicted and measured H…H couplings; it may simply be the model that the theory has been applied to is still incomplete but that the underlying quantum mechanics is not broken.
Posted on behalf of Bob Pascal.
=====
All of my previously reported NMR calculations were performed using the keywords “NMR=SPINSPIN”. At Dr. Rzepa’s request, I performed several more calculations using the Gaussian keywords “NMR=(SPINSPIN,MIXED)”. These reputedly give better estimates of scalar coupling constants, but they incur an enormously greater computational cost; thus, although the earlier calculations gave the coupling between all nuclei in the molecule, these new calculations were limited to the four axial atoms (Si1, H1, H2, and Si2), which, I suppose, are the only ones anyone cares about. The actual keywords were “NMR=(SPINSPIN,MIXED,READATOMS)”, with a list of the target atoms at the end of the input file.
The NMR data for compound 1 were recalculated in this manner at the B3LYP/6-31G(d), B3LYP/6-311G(d,p), and B3LYP/6-311+G(2d,p) levels. The resulting Si-H coupling constants were near to the experimental values, whereas the earlier calculations significantly underestimated these values. The results for the H…H coupling were mixed; the calculated coupling was reduced with the double zeta basis set, but increased with the triple zeta basis sets, both giving calculated coupling constants larger than the line widths of the singlets in the experimental spectrum.
We are thus no closer to a resolution of this problem.
We have just designed some new unsubstituted hydrocarbon structures with H…H contacts below 1.2 A. This is a new world record.
Check the communication in ChemPhysChem doi: 10.1002/cphc.201501002
http://onlinelibrary.wiley.com/doi/10.1002/cphc.201501002/abstract
I note here (DOI: 10.1002/anie.201712304) a new in,in cyclophane with two CH methine groups exhibiting a non-bonded H-H contact of 1.50-1.53Å and significantly a 1H coupling of 2.0 Hz. The 3D coordinates can be viewed at DOI 10.5517/ccdc.csd.cc1q9jbw.