Posts Tagged ‘animation’
Thursday, April 4th, 2019
Previously, I explored the Graham reaction to form a diazirine. The second phase of the reaction involved an Sn2′ displacement of N-Cl forming C-Cl. Here I ask how facile the simpler displacement of C-Cl by another chlorine might be and whether the mechanism is Sn2 or the alternative Sn1. The reason for posing this question is that as an Sn1 reaction, simply ionizing off the chlorine to form a diazacyclopropenium cation might be a very easy process. Why? Because the resulting cation is analogous to the cyclopropenium cation, famously proposed by Breslow as the first example of a 4n+2 aromatic ring for which the value of n is zero and not 1 as for benzene.[1] Another example of a famous “Sn1” reaction is the solvolysis of t-butyl chloride to form the very stable tertiary carbocation and chloride anion (except in fact that it is not an Sn1 reaction but an Sn2 one!)
The reason for posing this question is that as an Sn1 reaction, simply ionizing off the chlorine to form a diazacyclopropenium cation might be a very easy process. Why? Because the resulting cation is analogous to the cyclopropenium cation, famously proposed by Breslow as the first example of a 4n+2 aromatic ring for which the value of n is zero and not 1 as for benzene.[1] Another example of a famous “Sn1” reaction is the solvolysis of t-butyl chloride to form the very stable tertiary carbocation and chloride anion (except in fact that it is not an Sn1 reaction but an Sn2 one!)
(more…)
References
- R. Breslow, "SYNTHESIS OF THE s-TRIPHENYLCYCLOPROPENYL CATION", Journal of the American Chemical Society, vol. 79, pp. 5318-5318, 1957. https://doi.org/10.1021/ja01576a067
Tags:animation, Carbenium ion, Cations, Chemical elements, chemical reaction, Chemistry, Chlorine, computational chemistry, Cyclopropenium ion, Diazirine, energy, energy profile, free energy, Halogens, Natural sciences, Nucleophilic aromatic substitution, Oxidizing agents, Physical sciences, potential energy surface, SN1 reaction, Substitution reactions
Posted in reaction mechanism | No Comments »
Monday, February 18th, 2019
Students learning organic chemistry are often asked in examinations and tutorials to devise the mechanisms (as represented by curly arrows) for the core corpus of important reactions, with the purpose of learning skills that allow them to go on to improvise mechanisms for new reactions. A common question asked by students is how should such mechanisms be presented in an exam in order to gain full credit? Alternatively, is there a single correct mechanism for any given reaction? To which the lecturer or tutor will often respond that any reasonable mechanism will receive such credit. The implication is that a mechanism is “reasonable” if it “follows the rules”. The rules are rarely declared fully, but seem to be part of the absorbed but often mysterious skill acquired in learning the subject. These rules also include those governing how the curly arrows should be drawn.† Here I explore this topic using the Graham reaction.[1]‡
(more…)
References
- W.H. Graham, "The Halogenation of Amidines. I. Synthesis of 3-Halo- and Other Negatively Substituted Diazirines<sup>1</sup>", Journal of the American Chemical Society, vol. 87, pp. 4396-4397, 1965. https://doi.org/10.1021/ja00947a040
Tags:/RT, activation energy, activation free energy, animation, arrow pushing, arrow-head, cellular telephone, Chemical kinetics, chemical reaction, Chemistry, computed energy, Ed Smith, energy, energy maximum, energy minima, energy plot, energy profile, energy surface, free energy, lecturer, mechanism, Natural sciences, Organic chemistry, overall reaction energy, Physical sciences, Reaction rate constant, Resonance, Transition state, Transition state theory, tutor, Tutorial
Posted in Curly arrows, Interesting chemistry | No Comments »
Sunday, October 1st, 2017
I noted in my WATOC conference report a presentation describing the use of calculated reaction barriers (and derived rate constants) as mechanistic reality checks. Computations, it was claimed, have now reached a level of accuracy whereby a barrier calculated as being 6 kcal/mol too high can start ringing mechanistic alarm bells. So when I came across this article[1] in which calculated barriers for a dyotropic ring expansion observed under mild conditions in dichloromethane as solvent were used to make mechanistic inferences, I decided to explore the mechanism a bit further.
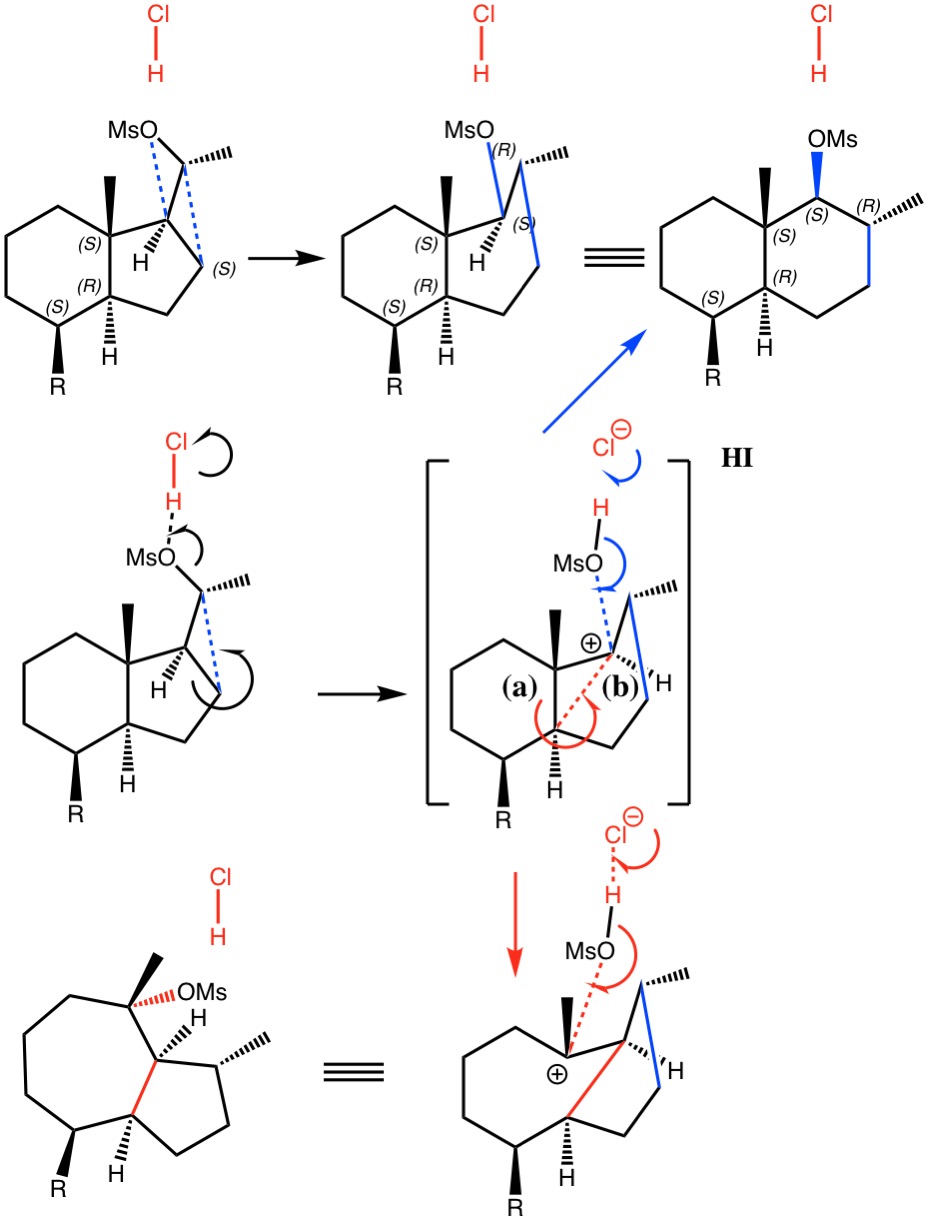
(more…)
References
- H. Santalla, O.N. Faza, G. Gómez, Y. Fall, and C. Silva López, "From Hydrindane to Decalin: A Mild Transformation through a Dyotropic Ring Expansion", Organic Letters, vol. 19, pp. 3648-3651, 2017. https://doi.org/10.1021/acs.orglett.7b01621
Tags:animation, bicyclic ring product, energy derivative gradient norm, energy profile, final non-ionic product, Organic chemistry, possible products, potential energy surface, realistic model for the reaction
Posted in pericyclic, reaction mechanism | 3 Comments »
Thursday, September 21st, 2017
A recent article reports, amongst other topics, a computationally modelled reaction involving the capture of molecular hydrogen using a substituted borane (X=N, Y=C).[1] The mechanism involves an initial equilibrium between React and Int1, followed by capture of the hydrogen by Int1 to form a 5-coordinate borane intermediate (Int2 below, as per Figure 11).‡ This was followed by assistance from a proximate basic nitrogen to complete the hydrogen capture via a TS involving H-H cleavage. The forward free energy barrier to capture was ~11 kcal/mol and ~4 kcal/mol in the reverse direction (relative to the species labelled Int1), both suitably low for reversible hydrogen capture. Here I explore a simple variation to this fascinating reaction.∞
(more…)
References
- L. Li, M. Lei, Y. Xie, H.F. Schaefer, B. Chen, and R. Hoffmann, "Stabilizing a different cyclooctatetraene stereoisomer", Proceedings of the National Academy of Sciences, vol. 114, pp. 9803-9808, 2017. https://doi.org/10.1073/pnas.1709586114
Tags:Ammonia borane, animation, Boranes, Chemistry, Cleaning Services, Company: React Group, free energy barrier, Hydroboration, Hydrogen, Matter
Posted in reaction mechanism | 1 Comment »
Thursday, May 25th, 2017
It is a sign of the times that one travels to a conference well-connected. By which I mean email is on a constant drip-feed, with venue organisers ensuring each delegate receives their WiFi password even before their room key. So whilst I was at a conference espousing the benefits of open science, a nice example of open collaboration was initiated as a result of a received email.‡
(more…)
Tags:animation, chemical reactions, City: Cupertino, Company: Cupertino Elec, Company: Firefox Communic, Computer Hardware - NEC, computing, detective, Digital media, Drip, Electronic documents, Electronic publishing, Email, HTML, Imperial College, Linux, operating system, Password, Person Location, Steven Kirk, Technology/Internet, XML
Posted in Chemical IT | No Comments »
Monday, March 20th, 2017
The example a few posts back of how methane might invert its configuration by transposing two hydrogen atoms illustrated the reaction mechanism by locating a transition state and following it down in energy using an intrinsic reaction coordinate (IRC). Here I explore an alternative method based instead on computing a molecular dynamics trajectory (MD).
(more…)
Tags:animation, chemical reaction, Chemistry, computational chemistry, computed potential energy surface, energy, Gaseous signaling molecules, Hydrogen, kinetic energy, kinetic energy contributions, Methane, Molecular dynamics, Physical chemistry, Quantum chemistry, Reaction coordinate, simulation, Theoretical chemistry
Posted in reaction mechanism | 2 Comments »
Monday, October 31st, 2016
Is asking a question such as “what is the smallest angle subtended at a chain of three connected 4-coordinate carbon atoms” just seeking another chemical record, or could it unearth interesting chemistry?
(more…)
Tags:animation, Bicyclic molecule, chemical record, Chemistry, City: Cambridge, Cycloalkane, Cyclopropanes, Java, Molecular geometry, Organic chemistry, potential energy surface, Safari, Web browser, X-ray
Posted in crystal_structure_mining, reaction mechanism | 7 Comments »
Saturday, November 28th, 2015
In answering tutorial problems, students often need skills in deciding how much time to spend on explaining what does not happen, as well as what does. Here I explore alternatives to the mechanism outlined in the previous post to see what computation has to say about what does (or might) not happen.
(more…)
Tags:animation, Chemical bond, condensation, Demjanov rearrangement, energy, Rearrangement reactions, Tiffeneau–Demjanov rearrangement
Posted in Interesting chemistry | No Comments »
Monday, November 23rd, 2015
This reaction emerged a few years ago (thanks Alan!) as a tutorial problem in organic chemistry, in which students had to devise a mechanism for the reaction and use this to predict the stereochemical outcome at the two chiral centres indicated with *. It originates in a brief report from R. B. Woodward’s group in 1973 describing a prostaglandin synthesis,[1] the stereochemical outcome being crucial. Here I take a look at this mechanism using computation.
(more…)
References
- R.B. Woodward, J. Gosteli, I. Ernest, R.J. Friary, G. Nestler, H. Raman, R. Sitrin, C. Suter, and J.K. Whitesell, "Novel synthesis of prostaglandin F2.alpha.", Journal of the American Chemical Society, vol. 95, pp. 6853-6855, 1973. https://doi.org/10.1021/ja00801a066
Tags:activation energy, animation, energy, learning tool
Posted in Interesting chemistry, reaction mechanism | 4 Comments »