This reaction emerged a few years ago (thanks Alan!) as a tutorial problem in organic chemistry, in which students had to devise a mechanism for the reaction and use this to predict the stereochemical outcome at the two chiral centres indicated with *. It originates in a brief report from R. B. Woodward’s group in 1973 describing a prostaglandin synthesis,[1] the stereochemical outcome being crucial. Here I take a look at this mechanism using computation.

The amino group is firstly converted to a diazonium chloride by nitrous acid and the resulting group is then easily eliminated. The problem is easy once you spot that either of the coloured bonds in the reactant is approximately antiperiplanar to the diazonium group, and might migrate to contract the ring. The green bond has a dihedral angle of ~174° with respect to the C-N≡N bond whilst the red bond has a less optimal value of ~166°. This alignment can also be viewed using orbital overlaps, in this case the (localised) NBO corresponding to the green or red bond and the empty antibonding NBO for the C-N bond. Below, the blue phase of the C-C bond is presumed to overlap constructively with the purple phase of the C-N anti bond, and likewise for the red/orange phases for the red bond.
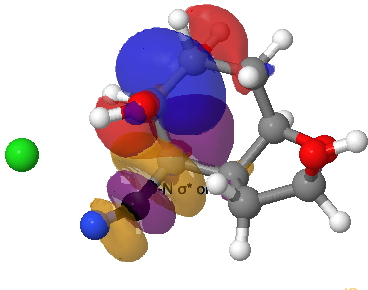
Click for 3D
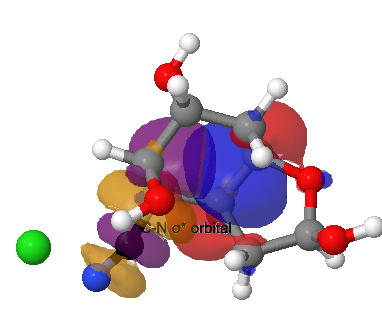
Click for 3D
A transition state (ωB97XD/6-311G(d,p)/SCRF=dichloromethane) can be located[2] and this yields[3] the reaction animation shown below;
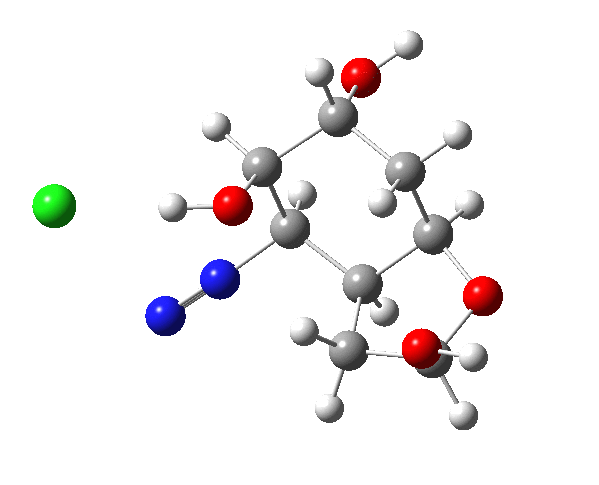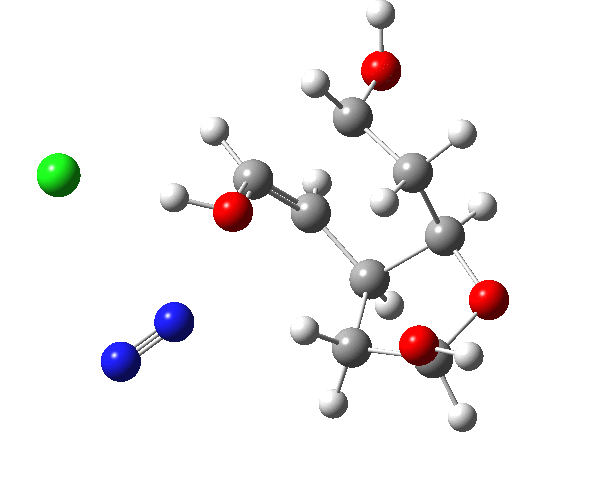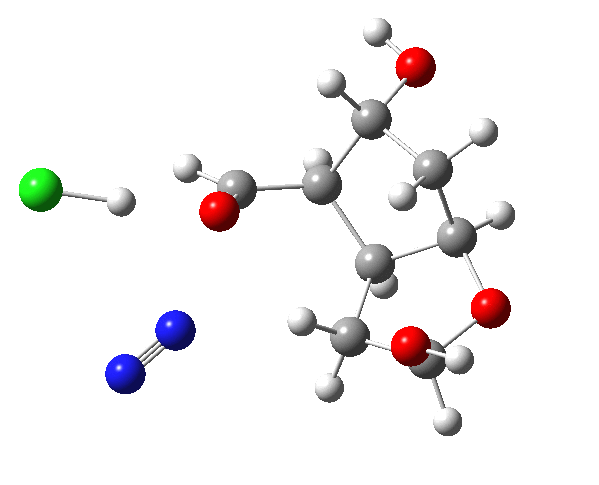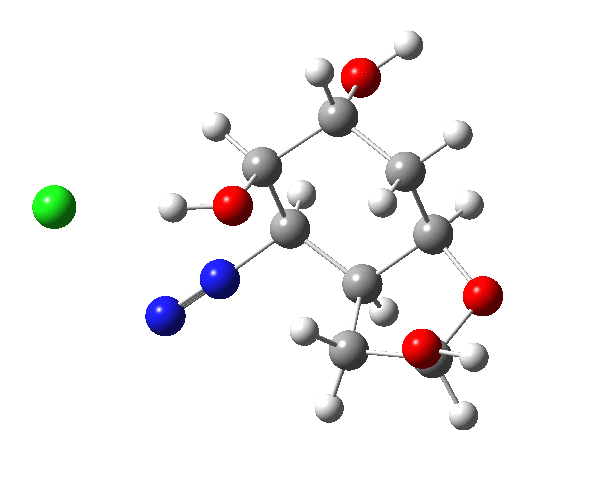
This has lots of interesting features, itemised below. The essence of the mechanism is that the green bond is induced to migrate by the proton removal from the OH bond by the chloride group. The red bond, although also aligned more or less correctly, has no such assistance.
- Plot 1 of energy shows a small activation energy (7 kcal/mol), leading to an exothermic reaction by about 34 kcal/mol.
- The gradient plot 2 (the derivative of the energy with respect to the geometry) shows several interesting features
- The reaction starts at IRC = 1.5 with zero gradients.
- It reaches the transition state very early (IRC=0.0), at which point the gradients are again zero.
- and then the gradients (almost but not quite) reach zero again (IRC ~-2). This is called a hidden reaction intermediate and corresponds to the cations noted above (as an ion pair, with chloride anion). Because the ion pair has a large dipole moment, one might expect the reaction to be sensitive to the polarity of any solvent, and these hidden intermediates might become real ones in highly polar solvents.
- At IRC -5, the gradients become large as the carbon starts to migrate.
- The migration (with retention of stereochemistry, it is a cationic [1,2] sigmatropic shift) is induced by the chloride anion starting to abstract the proton from the OH group, in synchrony with the carbon migration.
- After IRC -8, we see only conformational changes occurring, which may also be interesting to analyse.
- Plot 3 shows the length of the breaking (migrating) C-C (green) bond. It hardly changes up to the transition state; it is only afterwards that it really starts to break/migrate. Curiously, the red bond actually lengthens more than the green one at this stage (watch the animation above carefully) before changing its mind and reforming.
- Plot 4 the length of the newly forming (migrating) C-C bond. Note how initially, up to the transition state, this bond also lengthens (rather more than the green one does), before slowly reversing itself to contract at the transition state after IRC -3.
- Plots 5 and 6 show the lengths of the O…H and Cl…H bonds as the proton transfer proceeds. This mostly occurs AFTER the transition state is passed, and so the reaction should not exhibit any primary kinetic isotope effect induced by e.g. deuterium substitution.
- Plot 7 shows the dipole moment evolving along the reaction. At the start the species is an ion pair (diazonium chloride), but as the reaction proceeds HCl is formed and the dipole moment decreases to that of a less ionic compound.







As a learning tool, I find such animations carry a lot of information about reactions and their mechanism and it does not take more than a day or so to chart their course in the manner above.
Author
References
- R.B. Woodward, J. Gosteli, I. Ernest, R.J. Friary, G. Nestler, H. Raman, R. Sitrin, C. Suter, and J.K. Whitesell, "Novel synthesis of prostaglandin F2.alpha.", Journal of the American Chemical Society, vol. 95, pp. 6853-6855, 1973. https://doi.org/10.1021/ja00801a066
- H.S. Rzepa, "C 8 H 13 Cl 1 N 2 O 4", 2015. https://doi.org/10.14469/ch/191625
- H.S. Rzepa, "C8H13ClN2O4", 2015. https://doi.org/10.14469/ch/191626
Tags: activation energy, animation, energy, learning tool
I believe the hydroxy group next to the amino group in the starting material should be equatorial, not axial; i.e., inverted. At least that would be the case for compound X in Woodward’s paper, where amino and the first hydroxy are trans to each other, and thus both equatorial. It might not change much, however, apart from the aldehyde in the product being formed as a different conformer.
Interestingly, we use basically the same reaction as an example in a problem set on rearrangement reactions, but this time published by E. J. Corey: DOI: 10.1016/S0040-4039(01)96327-0
The paper contains a footnote thanking Woodward for “…outlining his approach to us in advance of publication.” Corey’s paper in Tetrahedron Lett. was received 25.5.1973 in the US, but “received in UK for publication” on 28.6.1973; Woodward’s in JACS received on 23.6.1973. Some interesting competition between those groups back then?
Thanks Lukas for the correction.
I re-ran the reaction with the other epimer, but you can hardly tell the profiles apart from the original.
It is certainly true that the Woodward-Corey debates are firmly part of the folklore of that time. Elsewhere on this blog, I recorded another reaction performed by Corey in 1963 to synthesize the sesquiterpene dihydrocostunolide. Indeed, I set this reaction as an exam question a few years ago. With this system, Corey came within a cat’s whisker of defining an iconic reaction that defined the pericyclic selection rules. Of course, it was Woodward a year or so later who did so, and his collaborator Hoffmann went on to win the Nobel prize for that work.
Ray Conrow has contacted me with another stereochemical correction based on Woodward’s article, being the endo/exo stereochemistry at the ring junction. The IRC for this isomer is very similar to those shown above. The data can be found at DOI: 10.14469/hpc/8067.
This looks great! Poetry in motion — so graceful. And the chloride ion saunters past, then swoops in to take the proton off to new adventures!