Recollect the suggestion that diazomethane has hypervalent character[1]. When I looked into this, I came to the conclusion that it probably was mildly hypervalent, but on carbon and not nitrogen. Here I try some variations with substituents to see what light if any this casts.

I have expanded the resonance forms of diazomethane by one structure from those shown in the previous two posts (a form by the way not considered in the original article[1]) to include a nitrene. This takes us back to an earlier suggestion on this blog that HC≡S≡CH is not a stable species but a higher order saddle point which distorts down to a bis-carbene, together with the suggestion that hypervalent triple bonds have the option of converting four of the six electrons into two carbene lone pairs, replacing the triple bond with a single bond. This in turn harks back to G. N. Lewis’ 101 year old idea for acetylene itself!
To explore this mode, I start by replacing the terminal ≡N in diazomethane with a ≡C-Me group, which cannot absorb electrons into lone-pairs in the manner that nitrogen can. A ωB97XD/Def2-TZVPP calculation‡ reveals that the linear form is a transition state for interconversion into a carbene. The IRC for the process (below) shows this carbene is ~10 kcal/mol lower than the linear “hypervalent” form.

NBO analysis of this transition state reveals a similar orbital pattern to diazomethane itself, including a non-bonding orbital on the H2C carbon. The Wiberg carbon bond indices are 3.6764 and N 3.6454 and the bond orders C=N 1.1390 and N=CMe 1.6192.
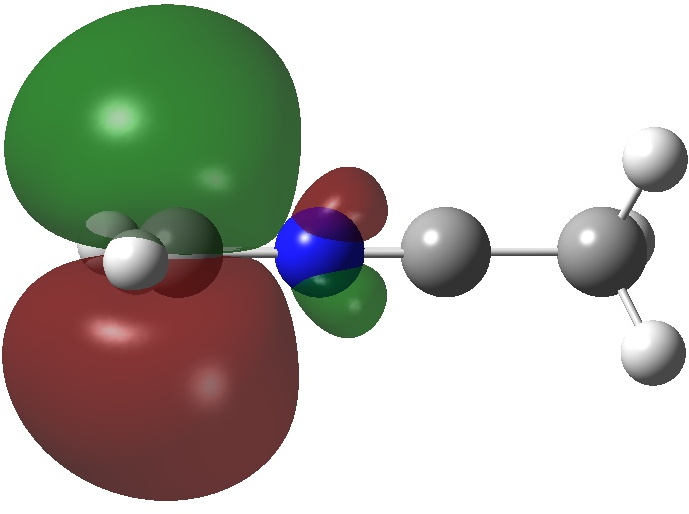
ELF analysis of this transition state reveals the presence of two non-bonding pairs on the carbon atoms either side of the nitrogen but unshared with it, with populations of 1.19e and 1.37e (DFT). That nitrogen really does not like excess electrons! The four atoms C,N,C,C have ELF valence basins totalling 8.00, 6.94, 7.69 and 7.92e (DFT) or 8.07, 7.07 and 7.61e (CASSCF), suggesting that unlike diazomethane itself, the octet-excess induced hypervalence on carbon is slightly decreased.
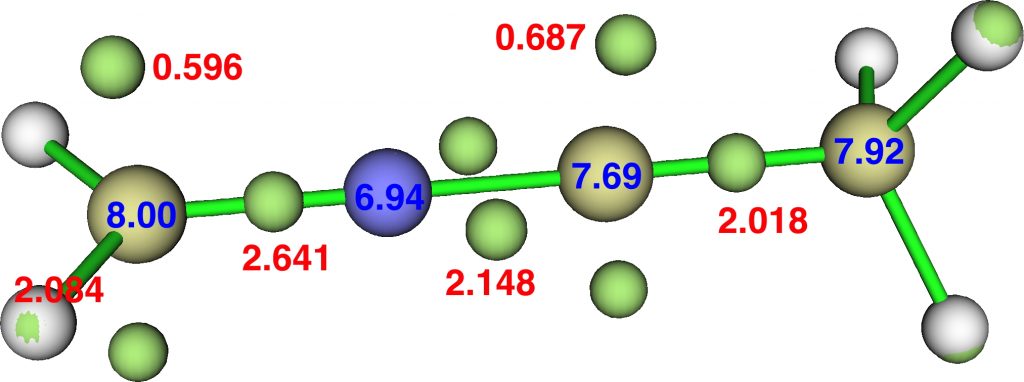
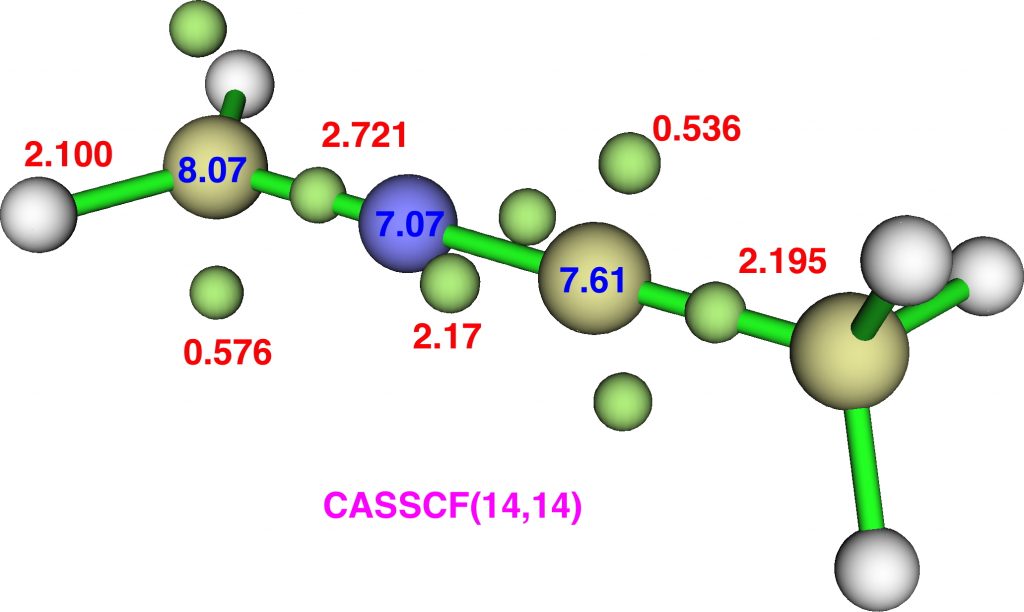
Pumping even more electrons in by replacing the ≡C-Me group with ≡C-NH2 does not increase any hypervalence, but does induce more electrons to reside in “lone pairs”. Of the four atoms along the chain, three have “lone pairs” associated with them, a total of 4.83e that do not contribute to bonds (valence).
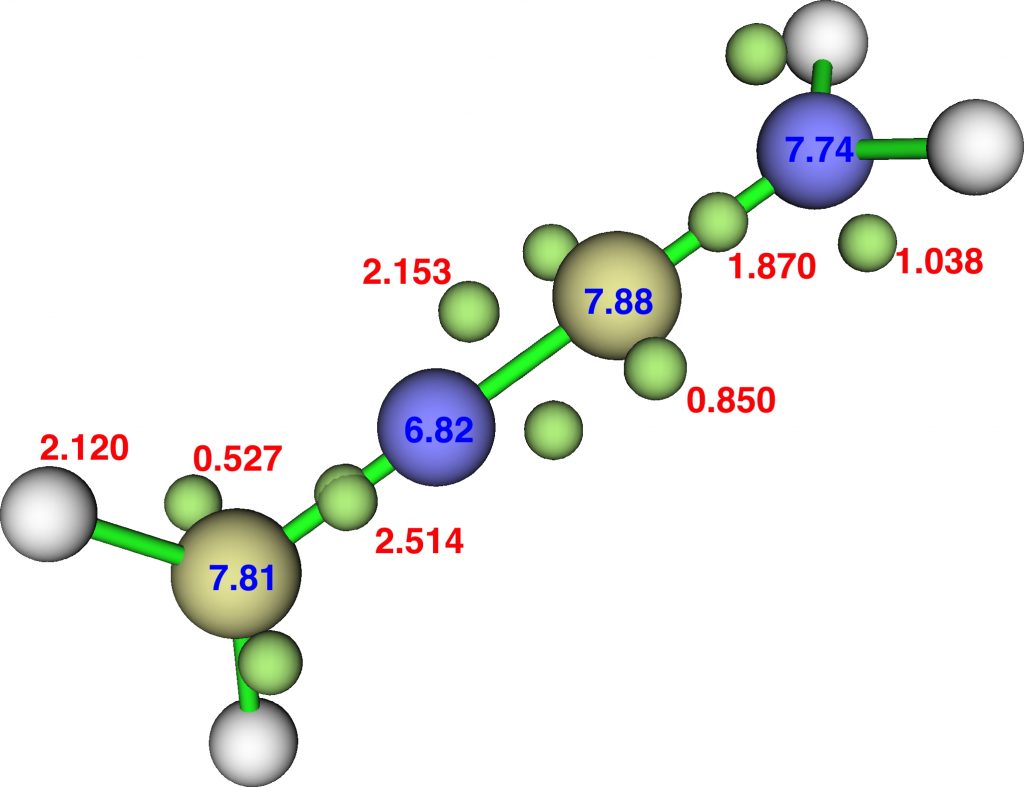
An electron withdrawing ≡C-CN group replacing the ≡C-NH2 reverses the effect of the latter, but this linear species is still a transition state for carbon isomerisation:
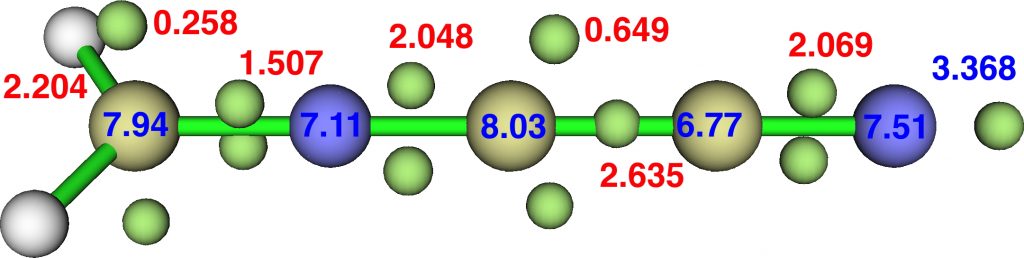
Finally, combining all we have learnt by adding in nitro groups on the first carbon. This is no longer a transition state but now a stable species; the sum of the ELF basin integrations around the carbon on the left reaches 8.95e, slightly higher than the dinitro-diazomethane discussed in the previous post. The numerical Wiberg atom bond indices are C 3.8713, N 3.6898, C 3.8503, C 3.9958 and N 3.0288 for the atoms along the chain, with the first nitrogen the “least-valent”.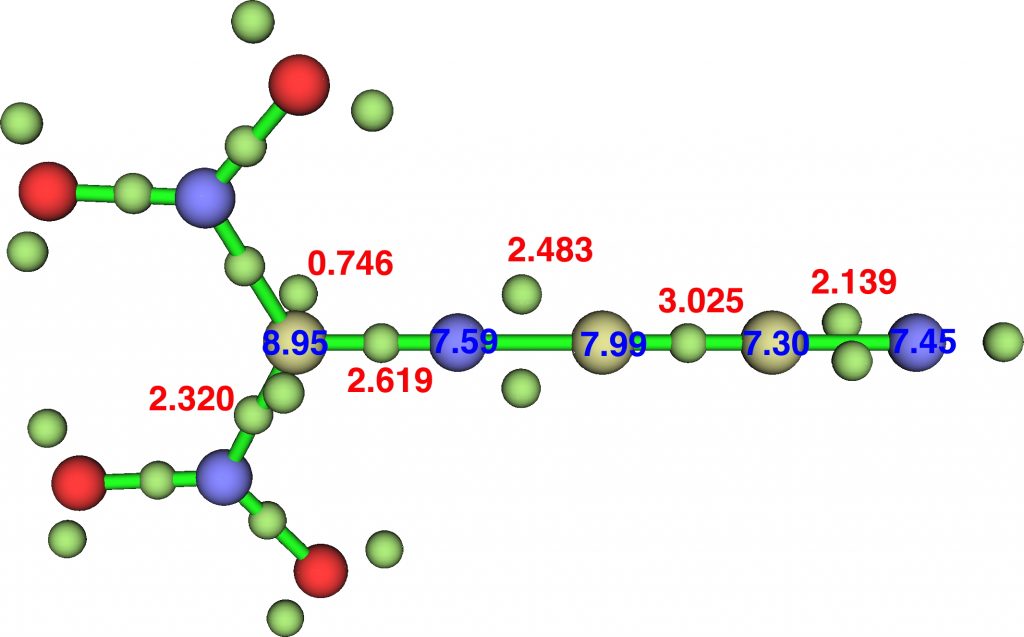
So we see that “hypervalence”, or at least “octet-excess”, which is not exactly the same as hypervalence since it includes contributions from non-bonding electrons, is balanced on a knife-edge. Trying to increase the octet-excess by pumping electrons in turns the system into a transition state for carbene formation. Octet-excess is seen as a metastable property, to be relieved by geometric distortions where possible or localization of electrons into non-bonding lone pairs. And I remind yet again that no evidence has manifested in calculations of the molecules above that the central nitrogen of these diazomethane-like systems has any propensity for octet or valence-excess as implied by the formula C=N≡X.[1]
‡FAIR data for all calculations is available at DOI: 10.14469/hpc/3476
Author
References
- M.C. Durrant, "A quantitative definition of hypervalency", Chemical Science, vol. 6, pp. 6614-6623, 2015. https://doi.org/10.1039/c5sc02076j
Tags: chemical bonding, Chemistry, diazo, Diazo compounds, Diazomethane, diazomethane-like systems, Functional groups, Hypervalent molecule, Molecular geometry, Organic chemistry, Recollects
I read the paper briefly and honestly it did not impress me at all. The reason is that a part of this so-called hyper-valency index is taken from quantum mechanical computations, ELF/AIM/NBO, but the other part is oversimplification of Lewis model. Why not taking everything from quantum mechanics?
I would replace the number of non-bonded electrons with AIM Localization index (LI). The number of bonded electrons for each atom would be half of delocalized electrons, i.e. half of the delocalization index(DI?2). If you sum up LI and DI/2, you will end up simply to the AIM charge. Will it exceed 8? Scarcely, to the best of my empirical knowledge.
This work is a good example of inventing a fashionable tool in chemistry that is of no or little use.
Re the oversimplification of the Lewis model. This does rather relate to eg Valence-bond theory where the selection of VB structures for the computation is crucial. If key structures are not included, you will not get sense out of the system. Thus the “nitrene” VB form, which like the “hypervalent” form carries no formal Lewis charges, was not considered in Durrant’s article.
The AIM localization index, all the required WFN (or FCHK) files are available via the FAIR data depositions. I will report back on LI + DI/2 shortly to see how these values compare with those inferred from the ELF integrations.
LI+1/2DI will produce exactly the AIM population. It seldom exceeds 8. Even in case of very electronegative-electropositive bonds poulation does not exceed 8. For instance in systems like LiF, NaF, or even CsF, F does not get more than 8 electrons. However, for some radicals like CaF, the picture might change. F may get more electrons than 8 in its valence shell in CaF radical. I do not expect I to see any violation of Lewis octet in diazomethanes.
As Durrant points out, an early GVB analysis of diazomethane (DOI: 10.1039/CS9972600087) proposed that “the central N atom takes part in five electron pair bonds“. Further, “despite the clear formation of five fully fledged electron-pair bonds, the overlaps between them are such that the average number of electrons around the nitrogen is very close to 7” and “the C atom is slightly negative. The central N atom is almost exactly neutral.“. This latter assertion is contrary to Durrant’s representation of the charge map for diazomethane (below, scheme 8 in the article) which appears to show the C atom as strongly positive and the central nitrogen strongly negative. Given that Durrant’s conclusions are based on such charge-maps, inferences from that charge map should be taken with some caution. As it happens, the NBO natural charges for my calculation show the carbon -0.502 (the hydrogens +0.230 each) the central nitrogen +0.07 and the terminal nitrogen -0.029, similar to the VB analysis.
Here is the relevant part of the DFT-based QTAIM analysis (the full analysis is now deposited with the FAIR data collection)
The charges on the C,N,N respectively are +0.38, -0.61, +0.08, which approximately matches Durrant’s diagram above. None of the atoms exceed the octet. These values are very different from the NBO scheme and the results inferred from ELF analysis. I guess we need to find out what the basis for this difference is.
I do not expect that ELF and NBO “populations” differ much from AIM population. I have this feeling that Durrant is twisting data in a way to interpret them in favour of his work. This is the base of my problem with this work. In fact, atomic population includes both bonded and non-bonded electrons this means that once count atomic population for charge, you already counted valence electrons. Durrant’s method is double-counting electrons that’s why you find hyper-valency.
If I was free enough, I would articulate my ideas about this work and write a comment on the paper. Unfortunately, I am too busy till April. If till that time no comment on the paper appears, I will write it!
P.S. It seems that WordPress has some issues. I do not receive notification for your comments on this post in spite of the fact that I asked to be notified and even received confirmation email.
I show below six valence bond structures for diazomethane. The top row were considered by Durrant. The bottom row includes a biradical, which has been shown to have a weight of 28% in VB structures (DOI: 10.1021/ja100512d). The weight of the nitrene form on the right is unknown, but it also has no formal charges.
Benoît Braïda (private communication with permission) sends me the results of some VBSCF calculations using a 6-31G(d) basis which indicate that the “nitrene” VB structure shown above contributes around 11% to the total ground state wavefunction, and 30% in the first excited state. Given that the contribution from the biradical is ~28%, already we see ~39% contribution from non-ionic structures. There are 18 VB structures in the complete set.