The potential energy surface for a molecule tells us about how it might react. These surfaces have been charted for thousands of reactions using quantum mechanics, and their basic features are thought to be well understood. Coming across an entirely new feature is rare. So what do you make of the following?

The reaction is shown above[1], and on the face of it, it looks like a normal pericyclic cascade. The standard mechanism inferred from simple mechanistic rules is to rewrite the ylid 1 as a carbene 2. This then undergoes a carbene insertion into an alkene to give 3, followed by an electrocyclic ring opening to give a presumed intermediate 4, and finally a [1,5] hydrogen migration ending in 5. Fairly uncontroversial stuff, you might think. The criterion is that it looks reasonable (each step has precedent).
The above reaction was discovered in 1992, before such simple mechanistic speculation could really be followed up by a good quality quantum mechanical investigation of how reasonable it really was. Well, this is 2014, and one need really spend no more than a few hours finding out. Before I present the results, it is worth reminding of the basic features of a potential energy surface:
- Reactant (intermediates) and products are all minima in such a surface. They are characterised by having 3N-6 (N= number of atoms) +ve force constants, and all the first derivatives (of geometric variables with respect to energy) are zero, as they also are for the next three types.
- A pair of minima can be connected by a transition state (a first order saddle point), for which 3N-7 of the force constants are +ve and precisely one is -ve.
- Less useful for mechanism are the 2nd order saddle points, which often connect a pair of transition states, and these have 3N-8 +ve force constants and 2 -ve ones. They are not kinetically important.
- Rarely, one finds two first order saddle points connected by a so-called valley ridge, and so one transition state can go downhill to another, and thence bifurcate into two possible products via a valley-ridge inflexion point.
- These four basic features have recently been augmented by so-called hidden intermediates. These emerge as a feature on the intrinsic reaction coordinate (IRC), being a frustrated minimum along that pathway. Frustrated, because the first derivatives never quite become zero (and the energy never quite a minimum) and so it does not qualify for any of the above definitions. Such points are increasingly being used to infer how small design changes to the reacting molecule might either fully stabilize such an intermediate, or perhaps remove it.
What we are about to discover relates to category 5, but with a new twist. Let us start with the IRC for 2 → 3[2] computed as ωB97XD/6-311G(d,p)/methanol.
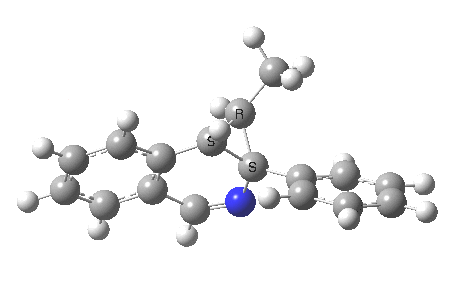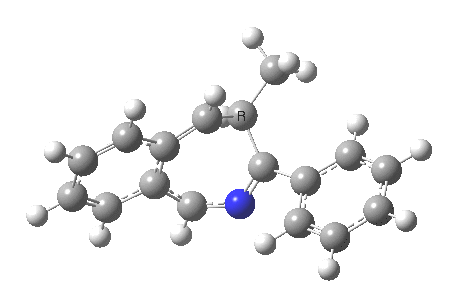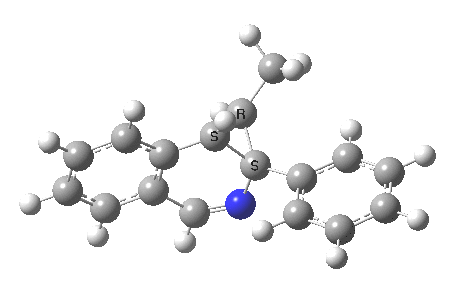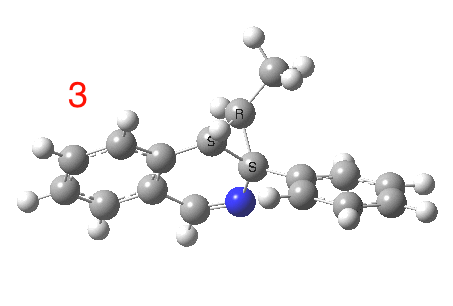


- The first noteworthy feature is that the activation energy for this reaction is tiny (~ 2 kcal/mol). The reactant 1 is in fact generated in situ from the imidoyl chloride and potassium t-butoxide; in effect it reacts as soon as it is formed!
- After this early transition state has passed, the reaction appears to pause at IRC ~3.6. This is a nice example of a hidden intermediate. You can see from the gradient plot of the IRC that the derivatives became small at this point, but do not quite become zero. I have set the relative energy to be zero at this point, for a reason which will soon become apparent. And then the reaction picks up again.
- In the IRC region 8-14, we get a conformational phenomenon, the slow rotation of the phenyl group.
So, the scheme at the top is not correct! Species 4 is reached BEFORE species 3, and 4 is a hidden rather than a real intermediate.
So to the next reaction, which is the [1,5] hydrogen shift.[3] This in fact starts where the last left off, at 3, and ends at 5. But again, 4 crops up as a hidden intermediate! It is common to BOTH reactions.‡ Two completely different types of reaction share 4 as a common hidden intermediate. Think of it as two flight paths intersecting at a common point in 3D space.


- This reaction can be thought of as a concerted pericyclic cascade. By this I mean two consecutive pericyclic processes, separated not so much by a real intermediate as a hidden one (4). A conjoined pericyclic if you will.
- A reality check. The original report says that the reaction of 2 → 3 occurs at 25°C, and 3 is fully characterised by NMR. The next phase, 3 → 5 only occurs at 70°C. The rate at which 2 → 3 forms must be determined by the rate of formation of 1/2 and not by the pathway shown above. Then the route for 3 → 5 crosses the route taken by 2 → 3 and proceeds on upwards to the transition state for [1,5] H transfer. One might argue that when the 3 → 5 journey has reached 4 it has two options; to continue on to 5, or to go on to 2. Another question might relate to the original journey of 2 → 3. When it reaches point 4, could it then take a sharp turn and instead head for 5 thus by-passing 3 entirely? Well no, because at this stage 3 is entirely downhill, where 5 needs some more climbing doing.
- The reality check has just one fly in the ointment; the barrier to the [1,5] shift is ~40 kcal/mol, about 15 kcal/mol too high to occur at 70C. It might be that instead we have a base-catalysed bimolecular deprotonation/reprotonation as a competing pathway.
Nonetheless, it would be interesting to act as an observer and stand at crossroads 4 watching molecules go by. Some are coming from 2 and headed for 3, some are coming from the other direction heading for 5. Each set has a sense of direction of where they are headed (and memory of where they have come from). You might spot where I am going with this; molecular dynamics! But 4 certainly is an interesting feature on the potential energy surface of this system, and not one I have ever seen before (indeed, has anyone seen similar?).
‡ Because 4 is not a stationary point in the potential surface (its gradients are not zero), it can only be characterised in the context of the IRC pathway. So its two manifestations in the two different IRCs are very similar, but are not identical.
Author
References
- K.R. Motion, I.R. Robertson, J.T. Sharp, and M.D. Walkinshaw, "Reactions of diene-conjugated 1,3-dipolar intermediates: the formation of cyclopropa[c]isoquinolines from benzonitrile o-alkenylbenzyl ylides and their rearrangements to benzazepines", Journal of the Chemical Society, Perkin Transactions 1, pp. 1709, 1992. https://doi.org/10.1039/p19920001709
- H.S. Rzepa, "Gaussian Job Archive for C17H15N", 2014. https://doi.org/10.6084/m9.figshare.936551
- H.S. Rzepa, "Gaussian Job Archive for C17H15N", 2014. https://doi.org/10.6084/m9.figshare.936657
Tags: potential energy surface
Hi Henry, Fascinating! We’ve observed some similar things in carbocation rearrangements. See, for example, the IRCs in DOI: 10.1021/ja9084786 — note the B-like structures. Best, Dean
Hello Henry,
very enjoyable read, especially with the GIF movies, they really make a connection between the 2D chemical reaction drawing and the 3D real world. I foresee/hope that electronic chemistry books will have that functionality in the future, especially when discussing chemical reactions.
I wonder if the selection of the ωB97XD/6-311G(d,p) functional was a rationale decision, based on accuracy and computational cost. I would be interested what the reason was, potentially with some benchmarks or pointers to some references. Basically why not M11-L or SOGGA11X or N12-SX (or any others)? I seems there are still many unresolved challenges in reaction modelling, even in 2014 🙂
Thank you
Tobias
Yes, figures 2, 3 and 8(?) shows signs of hidden intermediates Dean. The gradient norms show this up even better!
Figure 8 in particular had difficulties in running the whole duration in one go; Gaussian 09 is so much better at this sort of thing. Worth re-running?
I note two more aspects
1. These are all cations; does solvation influence the IRC?
2. As cations, they all lack any counterion. Depending on the nature of the latter of course, would its presence also influence the IRC?
Tobias: The selection of a functional nowadays from the pool of 200+ available tends to be a combination of previous experience and some design. Thus if an IRC is envisaged, to get the outer behaviour correct, one really does need dispersion corrections. The ωB97XD has these, but it was also developed (parametrised) against reaction barriers, and there are a number of careful calibrations of its energies vs CCSD(T). It normally takes ~5 years for these calibrations to appear, and it may be sound advice not to use eg the newer functional unless one has access to such calibrations. In our case, we have not yet found ωB97XD wanting (we have many such calibrations in house), and so would jump ship from it only if a new functional could be demonstrated to be much better (or faster, or both). We have in fact recently established that eg the TZVP basis (about the same size as 6-311G(d,p)), has much smaller basis-set-superposition-errors, and so in some circumstances might be a better basis.
We do have an article hopefully coming out soon that actually goes over a lot of these decisions, so watch this space.
Tobias: re “especially with the GIF movies“. I have incorporated lots of such movies, orbitals, NCI surfaces and other “3D” components into my current lecture notes, along with much effort to ensure all of this can be viewed on tablets as well as conventional computers. See doi: 10042/a3uy9.
This collection can be converted into a “text book” via the ePub3 format, and then loaded like any other e-book into e.g. a Tablet. Or of course the tablet can just access the same via the HTML5/CCS3/Javascript compatible Web page above. Along with a 4th standard (SVG) this combination is certainly a highly viable way of authoring an electronic text book nowadays. Indeed, one might argue that the era of Office/PDF combinations, so ubiquitous for authoring nowadays, is rapidly being joined by the “HSCJ” combination above as something aspiring (electronic) text book authors should become familiar with. I have written a manifesto espousing this approach which you can read at doi: 10042/a3uz1 Comments welcome! And if you are baffled, do consult the “help” section of the notes where some of the issues are set out.
Oh, and I should add that these notes (text-book) are fully integrated into digital repositories, so that the reader can quickly acquire the original data used to create the diagrams or compose the narrative.
If anyone is attending the Dallas ACS meeting, I will be talking about some of these themes on the Monday afternoon.
Dear Henry,
For the based promoted 5-endo cyclization of benzyl alkynyl sulfides. Our gas phase calcs favored Baldwin product, but experiment contradicts this.
We used CPCM-CAM-B3LYP and we saw similar hidden intermediates (did not report it). At the time we thought it could be indicative of a valley ridge, but did not explore it further. We modeled required base MeOK explicitly.
Best,
Álvaro
IRC calculations were coded decades ago, but as they require constant derivative calculation and not infrequent 2nd derivative calculation, they were really only ever used in the early days to verify the presumed identity of a reactant and product connected by a transition state. Other than this information, they were not thought to be interesting in their own right so to speak.
With improvements in the calculation of 2nd derivatives, the increase in computing power, and improved solvation algorithms, IRCs have taken on a new lease of life. In some measure this has come about because one now has the luxury of computing many more points. Typically, this has increased from ~ 50 to 250 for each side of the transition state. The calculation also needs to be done at high accuracy (ultrafine grids for DFT).
Secondly, it is well worth inspecting the gradient norm along the pathway, since this reveals features easily overlooked from just the energy plot.
One of my favourites by the way remains the SN1 solvolysis of tert-butyl chloride in water where the carbocation, often regarded as a proper intermediate in the reaction, then emerges as a hidden intermediate. I also found hidden intermediates in 6-endo-dig Baldwin ring closures.
Hi,
I checked the courses at doi: 10042/a3uy9 its impressive to see such data working on an Iphone. The navigation is a bit combersome, but well thats probably a space issue.
Regarding the epub3 and organic and computational chemistry publishing manifesto doi: 10042/a3uz1 I think the technical barriers are still too high for the majority of researchers. Even capturing such content from Gaussian or JMOL requires some hacks and conversions, maybe as small as executing a script on the command line. Throwing it into a flat PDF is much easier. As long as publishers are not on the same boat, and easy templates are developed it will be a small group of tinkerers that push the field forward. Maybe exposing such content on a much wider scale, such as open electronic textbooks (http://chemwiki.ucdavis.edu/) can teach students that there is a better vibrant 3D world out there instead of the 2D dogmatic view.
Cheers
Tobias
Tobias: Re technical barriers to creating epub3 content. Yes, when Apple introduced the iBooks author as a really easy way of creating similar content, we thought the prospects might be good. But its a very bespoke system, and does not produce generic epub3 as far as we can see. There is some prospect of creating useful templates, and as browser support for the HTML5 element <link rel=”import” href=”template.html”> improves (only Google Chrome supports it at the moment) we might expect things to get a lot easier. All of the frightening stuff can be put into the template, leaving the researcher to concentrate on their own content.
“Throwing it into a flat PDF is much easier”. Creating PDF is normally simply “save as PDF” and there is no reason why “Save as epub3” might not become as familiar! Although I have not tried it, Adobe In-Design is supposed to be that easy (Adobe, the custodians of course also of the PDF).
Dean: “note the B-like structures“. I thought I might repeat Dean’s “C2-to-E ” reaction (Figure 4 in his article). Below is the result of a single run, covering 184 points along the IRC. I have included the gradient norm plot, which shows very clearly a “hidden intermediate” either side of the transition state. Calculation doi: 10.6084/m9.figshare.938192.
These features are of course due to the attempted formation of non-classical carbocations.
Tobias: “Maybe exposing such content on a much wider scale, such as open electronic textbooks (http://chemwiki.ucdavis.edu/) ”
I took a very quick look at chemwiki.ucdavis.edu/
Organic_Chemistry/Pericyclic_Reactions/Cycloaddition_Reactions which is the equivalent in terms of content of 10042/a3uy9. It is conventional in the sense that it could easily be a PDF file, and it does not contain any animations, transition states, orbitals or other 3D content.
Our own Wiki (also named ChemWiki when we set it up in March 2006) was from the outset enabled to show 3D content, and we use it still to teach various aspects of molecular modelling. It will shortly be refactored to also support tablets (they were not around in 2006!). And it has always been open!
I might finally add that we too are discussing MOOCs here. I think the phrase is distinctive contributions to the genre. Thus if you compare my take on pericyclic reactions and Bill Reusch’s, I think you will find them very very different. Dare I say, each is distinctive! I would not want the MOOC phenomenon to end up with the concept that each topic can/should be covered once in a global sense, and then all students are sent off to that one instance to “learn” about any particular topic. Diversity is good for science and good for chemistry. Pedagogy is all about assimilating the diversity and drawing your own conclusions from it. In that sense, I am not warming to MOOCS; they may simply homogenise, remove diversity, and cheapen everything. But those of us not charged with being the custodians of say the global MOOC on pericyclic reactions must redouble our efforts to be distinctive.
I set 10042/a3uy9 up very much with that in mind.