The outcome of pericyclic reactions con depend most simply on three conditions, any two of which determine the third. Whether the catalyst is Δ or hν (heat or light), the topology determining any stereochemistry and the participating electron count (4n+2/4n). It is always neat to conjure up a simple switch to toggle these; heat or light is simple, but what are the options for toggling the electron count? Here is one I have contrived by playing a game with the periodic table.  The ring closure of a divinylketone is called the Nazarov reaction, it being promoted thermodynamically by coordination of a Lewis acid to atom X. Divinyl ketone can be regarded as a hidden pentadienyl cation, since the C=O bond is polarised Cδ+Oδ- in the time-honoured manner of organic chemistry. In this (formal) resonance form, it becomes part of a pentadienyl cation and can electrocyclise via a 4-electron reaction involving a stereochemical process known as conrotation. The new bond is formed antarafacially (from opposite faces) at the termini of the pentadienyl cation (ωB97XD/6-311G(d,p)/SCRF=dichloromethane.[cite]10.6084/m9.figshare.1125721[/cite]). Note that for the uncatalysed reaction, the barrier is high and the reaction is endothermic but adding a BF3 to the oxygen lowers the barrier and removes the endothermicity.[cite]10.6084/m9.figshare.1125724[/cite]
The ring closure of a divinylketone is called the Nazarov reaction, it being promoted thermodynamically by coordination of a Lewis acid to atom X. Divinyl ketone can be regarded as a hidden pentadienyl cation, since the C=O bond is polarised Cδ+Oδ- in the time-honoured manner of organic chemistry. In this (formal) resonance form, it becomes part of a pentadienyl cation and can electrocyclise via a 4-electron reaction involving a stereochemical process known as conrotation. The new bond is formed antarafacially (from opposite faces) at the termini of the pentadienyl cation (ωB97XD/6-311G(d,p)/SCRF=dichloromethane.[cite]10.6084/m9.figshare.1125721[/cite]). Note that for the uncatalysed reaction, the barrier is high and the reaction is endothermic but adding a BF3 to the oxygen lowers the barrier and removes the endothermicity.[cite]10.6084/m9.figshare.1125724[/cite] 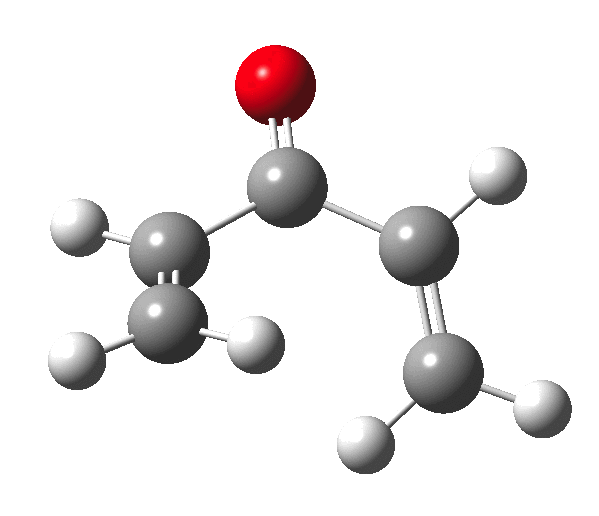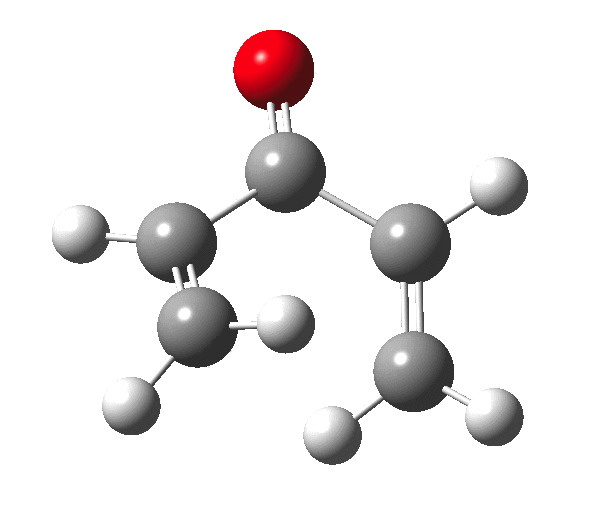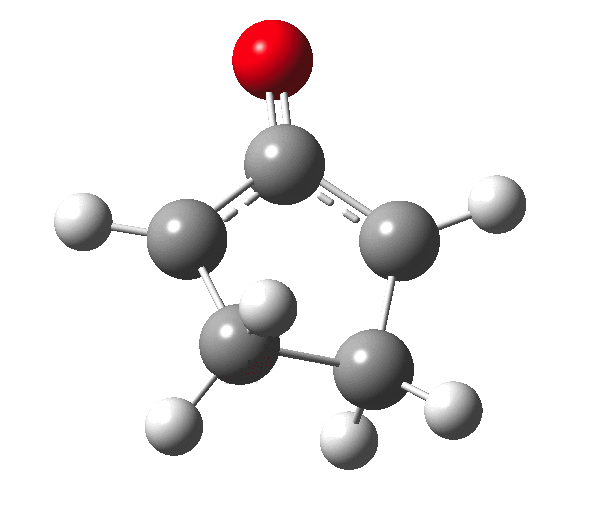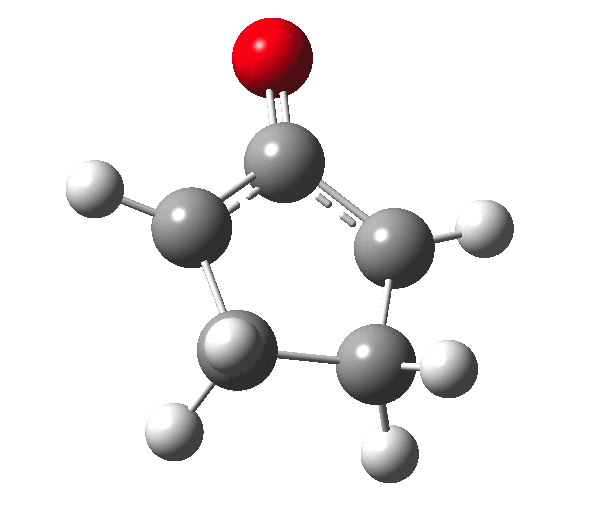

 So, one can play a game and ask what would happen if the polarity of the C=X bond were to be reversed. This means going left of oxygen in the periodic table, ending at Be.[cite]10.6084/m9.figshare.1125792[/cite] The reaction has a high barrier, but it is strongly exothermic.† However the most noteworthy aspect is that the stereochemistry of the electrocyclisation is now disrotatory, with suprafacial bond formation (from the bottom face in the animation below). The stereochemical outcome of this reaction has been flipped by reversing the polarity of the CX bond.‡
So, one can play a game and ask what would happen if the polarity of the C=X bond were to be reversed. This means going left of oxygen in the periodic table, ending at Be.[cite]10.6084/m9.figshare.1125792[/cite] The reaction has a high barrier, but it is strongly exothermic.† However the most noteworthy aspect is that the stereochemistry of the electrocyclisation is now disrotatory, with suprafacial bond formation (from the bottom face in the animation below). The stereochemical outcome of this reaction has been flipped by reversing the polarity of the CX bond.‡ 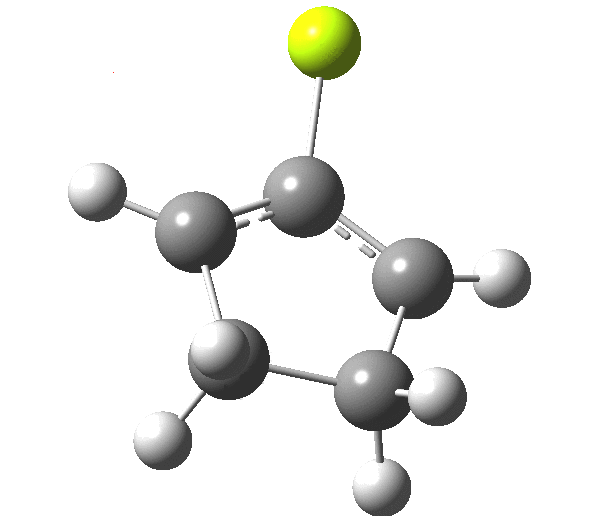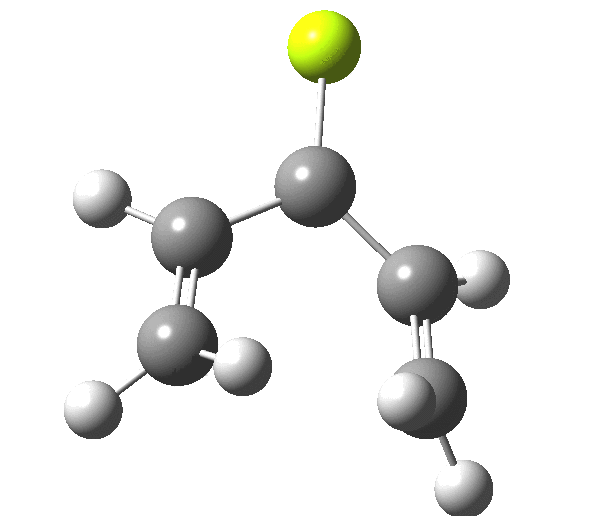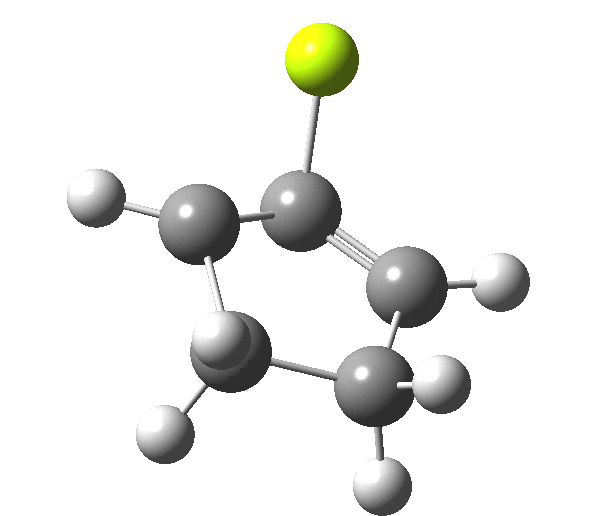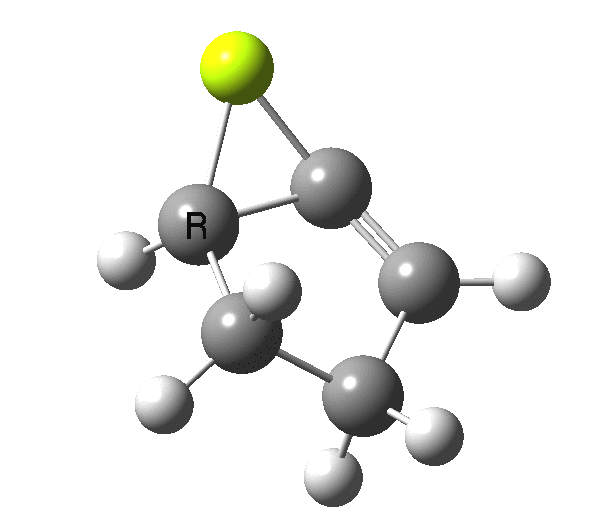
 This little example shows how a thought game played using the periodic table can then be reality tested by solving appropriate quantum mechanical equations. In this instance, one is not going to rush into the laboratory to try to replicate the experiment, but it might help catalyse new thoughts amongst the readers of this blog.
This little example shows how a thought game played using the periodic table can then be reality tested by solving appropriate quantum mechanical equations. In this instance, one is not going to rush into the laboratory to try to replicate the experiment, but it might help catalyse new thoughts amongst the readers of this blog.
Posts Tagged ‘animation’
Using a polar bond to flip the (stereochemical) outcome of a pericyclic reaction.
Monday, August 4th, 2014Why is the Sharpless epoxidation enantioselective? Part 1: a simple model.
Sunday, December 9th, 2012Sharpless epoxidation converts a prochiral allylic alcohol into the corresponding chiral epoxide with > 90% enantiomeric excess[cite]10.1021/jo00369a032[/cite],[cite]10.1021/jo00360a058[/cite]. Here is the first step in trying to explain how this magic is achieved.
Molecular gymnastics in 2+2 cycloadditions.
Wednesday, December 14th, 2011In this earlier post, I described how the stereochemistry of π2+π2 cycloadditions occurs suprafacially if induced by light, and how one antarafacial component appears if the reaction is induced by heat alone. I also noted how Woodward and Hoffmann (WH) explained that violations to their rules were avoided by mandating a change in mechanism requiring stepwise pathways with intermediates along the route. Here I illustrate how the stereochemistry of a thermal π2+π2 cycloaddition can indeed avoid an antarafacial component by performing appropriate gymnastic contortions instead of a mechanistic change (a WH violation certainly in the letter of their law, if not their spirit).
 (more…)
(more…)
The chemistry behind a molecular motor. The four wheels?
Friday, November 25th, 2011In the previous post, I wrote about the processes that might be involved in a molecular wheel rotating. A nano car has four wheels, and surely the most amazing thing is how the wheels manage to move in synchrony. This is one hell of a tough problem, and I do not attempt an answer here, but simply record an odd observation.
Atropisomerism in Taxol. An apparently simple bond rotation?
Tuesday, November 1st, 2011My previous post introduced the interesting guts of taxol. Two different isomers can exist, and these are called atropisomers; one has the carbonyl group pointing up, the other down. The barrier to their interconversion in this case is generated by a rotation about the two single bonds connecting the carbonyl group to the rest of the molecule. Introductory chemistry tells us that the barrier for rotation about such single bonds is low (i.e. fast at room temperature). But is that true here?
Mechanism of the reduction of a carboxylic acid by borane: revisited and revised.
Sunday, October 16th, 2011I asked a while back whether blogs could be considered a serious form of scholarly scientific communication (and so has Peter Murray-Rust more recently). A case for doing so might be my post of about a year ago, addressing why borane reduces a carboxylic acid, but not its ester, where I suggested a possible mechanism. Well, colleagues have raised some interesting questions, both on the blog itself and more silently by email to me. As a result, I have tried to address some of these questions, and accordingly my original scheme needs some revision! This sort of iterative process of getting to the truth with the help of the community (a kind of crowd-sourced chemistry) is where I feel blogs do have a genuine role to play.

The reduction of a carboxylic acid by borane
Less is more: the dyotropic rearrangement of ethane
Saturday, June 11th, 2011In a time when large (molecules) are considered beautiful (or the corollary that beauty must be big), it is good to reflect that small molecules may teach us something as well. Take ethane. Is there anything left which has not been said about it already? Well, consider the reaction below, in which two hydrogen atoms mutually hop from one carbon to the other.