The so-called E2 elimination mechanism is another one of those mainstays of organic chemistry. It is important because it introduces the principle that anti-periplanarity of the reacting atoms is favoured over other orientations such as the syn-periplanar form; Barton used this principle to great effect in developing the theory of conformational analysis. Here I explore its origins.
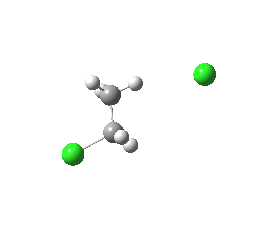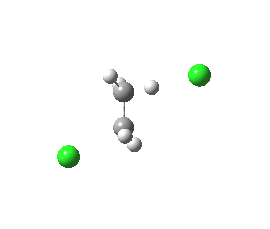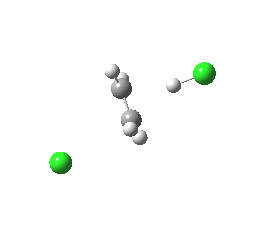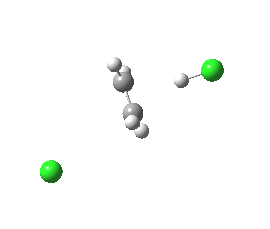
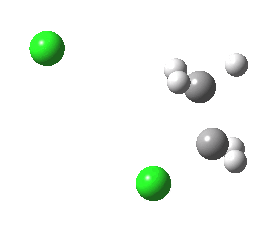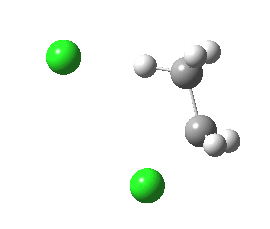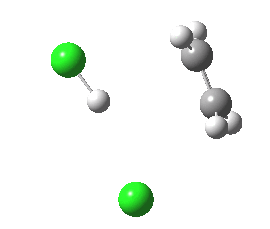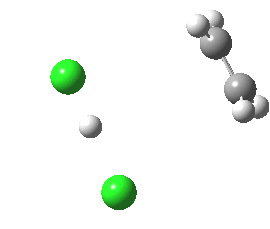
The transition state for this reaction is readily calculated (ωB97XD/6-311+G(d,p)/SCRF=acetone). The app form is calculated to be 4.3 kcal/mol lower in free energy than the spp form.
| Anti-periplanar | Syn-periplanar |
|---|---|
antiperiplanar E2 transition state. Click for 3D. |
syn-periplanar E2 transition state. Click for 3D. |
The concerted bimolecular nature of the elimination (E2) is revealed by the intrinsic reaction coordinate (IRC). Notice the peculiar goings on for the spp geometry at IRC = -15, during which the initial gauche conformation of the chloroethane rotates into an eclipsed orientation to allow the synperiplanar transition state to develop. The app isomer has no need to do this.
Time to explain what is going on. This will be done by obtaining a form of localised bond orbital known as the NBO. In fact, a pair of them, one bonding, the other antibonding. The former is known as the donor, and the latter the acceptor. Such a take was used in an earlier post to explain why 1,2-difluoroethane adopts a gauche rather than antiperiplanar conformation. Such localised orbitals are useful to probe specific bonds in a molecule, and why they might be reacting in the way that they do. The theory behind this is shown below. Basically an occupied (donor) orbital can interact (be perturbed by) an empty (acceptor) orbital, the result being a stabilisation energy E2. This energy depends on two specific factors; the energy gap between the donor and acceptor orbitals (the smaller the gap the better) and the overlap between the two orbitals (the larger the overlap the better).
For chloroethane, these two criteria are satisfied by virtue of the C-Cl bond being a good acceptor (due to the electronegative chlorine) whilst the C-H bond is a reasonable donor. The overlap is shown below (click on the image to get a rotatable model). The colour scheme is purple/orange for the two phases of the donor orbital, which derives from the H-C bond, and blue/red for the acceptor orbital, which derives from the C-Cl bond. The colours can be equivalenced; purple=blue, and orange=red, meaning that any overlap between purple and blue or orange and red is positive and stabilising, and the larger this overlap, the larger is E2. At the start of the reaction (IRC=9), E2 = 7.0 cal/mol.
Donor-acceptor interaction in the E2 reactants. Click for 3D.
Donor-acceptor interaction in the app E2 transition state. Click for 3D. |
Donor-acceptor interaction in the spp E2 transition state. Click for 3D. |
The syn-periplanar transition state in contrast shows a smaller value of E2 = 63 kcal/mol at the transition state due to a smaller positive overlap of the donor/acceptor orbitals at this geometry.
To conclude, the relatively small and subtle bond/antibond orbital overlaps that are used in conformational analysis to explain the equilibrium conformations of species such as 1,2-difluoroethane also apply to considering the transition states involving elimination. The overlap of the donor orbital (with two electrons) with an (empty) acceptor orbital directly morphs as the reaction proceeds into a new orbital in the product.
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
View Comments
This helped me so very much. Thanks for the write-up!
Hey Henry, Your job is amazing! Could you bring to me the IRC coordinates file?
The IRC is hyperlinked to the log file containing it above, as per http://dx.doi.org/10.14469/ch/11908