The properties of electrons are studied by both chemists and physicists. At the boundaries of these two disciplines, sometimes interesting differences in interpretation emerge. One of the most controversial is that due to Bader (for a recent review, see DOI: 10.1021/jp102748b) a physicist who brought the mathematical rigor of electronic topology to bear upon molecules. The title of his review is revealing: “Definition of Molecular Structure: By Choice or by Appeal to Observation?”. He argues that electron density is observable, and that what chemists call a bond should be defined by that observable (with the implication that chemists instead often resort to arbitrary choice). Here I explore one molecule which could be said to be the focus of the differences between physics and chemistry; cis-but-2-ene.
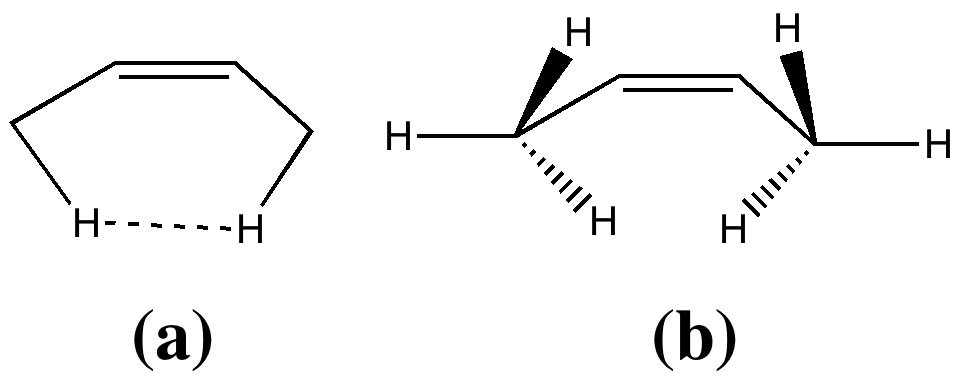
The structure of this system has been determined by electron diffraction to exhibit a H…H distance of ~2.1Å as in (a), DOI: 10.3891/acta.chem.scand.24-0043. Why is this of interest? Because a rotational alternative, shown as (b) could result in a significantly longer H…H distance (~ 2.6Å). Now bear in mind that the van der Waals radius of hydrogen is estimated at ~1.2Å, and that two hydrogens will be most strongly attracted by dispersion forces when separated by ~2.4Å. As they get closer, that attraction will be counterbalanced by a repulsion, which will eventually win out. Structure (b) does not benefit from H…H dispersion attractions, but are the hydrogens in structure (a) too close to do so as well?
Well, let us adopt Bader’s approach, and look at the topology (QTAIM) of the electronic distribution in structure (a). The features to concentrate on are the purple dots, which in this analysis have been named bond critical points (BCP) and the single yellow sphere, which is a ring critical point (RCP). There are two other types of critical points, which I will call nuclear attractors (NACP) and cage points (CCP). The total occurrences of all these critical points is determined by a topological theorem (Poincare-Hopf), which states that NACP-BCP+RCP-CCP=1. You can see below that the H…H region is indeed connected by a bond critical point (none of the other purple dots are controversial). To a physicist, this is a real feature of the (observable or in this instance the calculated) electron density. In effect, it is a topological bond. Unfortunately, chemists like to think of bonds as entities which result in stability; a bond contributes to the stability of a molecule (and an anti-bond, should such exist, to instability). Hence aromaticity (=stability) vs anti-aromaticity (=instability). Chemists, by choice, might prefer not to call the H…H region a bond because it is probably not contributing to the stability of the molecule. Of course, the two camps are arguing about different things; the topology of the electron density ρ(r) vs the energy of the molecule.
QTAIM topological analysis of cis but-2-ene. Click for 3D.
I have done so below using the NCI (non-covalent interaction) procedure (which I have commented on in many other posts here). The NCI surface is shown below, embedded within the NCI surface as purple dots are the BCP and RCP discussed above. Now, the NCI method cleverly attempts to ascertain whether a region is attractive or repulsive, and it colour codes the surface accordingly. In the below, blue is deemed attractive, and red repulsive. We immediately see that the H…H BCP is embedded in an attractive region, but the adjacent RCP is embedded in a repulsive region. Whatever attraction the BCP might be experiencing is negated by the repulsion the RCP has. The two cancel (annihilate). Taken together, the H…H region is probably repulsive (ways of quantifying how NCI regions integrate are being investigated and will be discussed in a future post). Perhaps this manner of looking at it satisfies both the physicists and the chemists?
The NCI surface for cis but-2-ene. Click for 3D.
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
View Comments
What is the result of NCI if the integration is done over the whole hydrogen basin
My suggestion is about the evaluation of NCI values through the
whole Hydrogen basin instead of just small portion of space around BCP
and RCP. Then we will have the complete picture (in NCI approach)
about the sign of potential energy (positive or negative). In other
words we will see whether the whole hydrogen basin lies in the
repulsion (Red) or attraction (Blue) part of potential energy in NCI
approach.
Interesting to see that NCI shows location of H-H BCP in the
attraction part of the potential energy,However I did not understand
why it has been concluded that the H-H contact is repulsive.
It is very interesting you ask about the whole H basin. We have done such an integration, according to the protocol described in DOI: 10.1021/jp204278k. The integration for the repulsive part appears bigger than the attractive part, although aspects still need sorting. There is also another post on this theme here which elaborates using an NBO argument.
Very interesting post, although I think that would not be reduced to a physicists vs. chemists issue. Indeed, Bader was an experimental physical organic chemists before moving to quantum and theoretical chemistry, why call him a physicist?
IHMO, the main criticism to Bader concept of bonds (chemical or not) is that relies on a 'topological' partition of molecular density. As a consequence his concept of an atom in the molecule is, in general, not a transferable entity, contrary to traditional chemical thinking.