Do you fancy a story going from simplicity to complexity, if not absurdity, in three easy steps? Read on! The following problem appears in one of our (past) examination questions in introductory organic chemistry. From relatively mundane beginnings, one can rapidly find oneself in very unexpected territory.
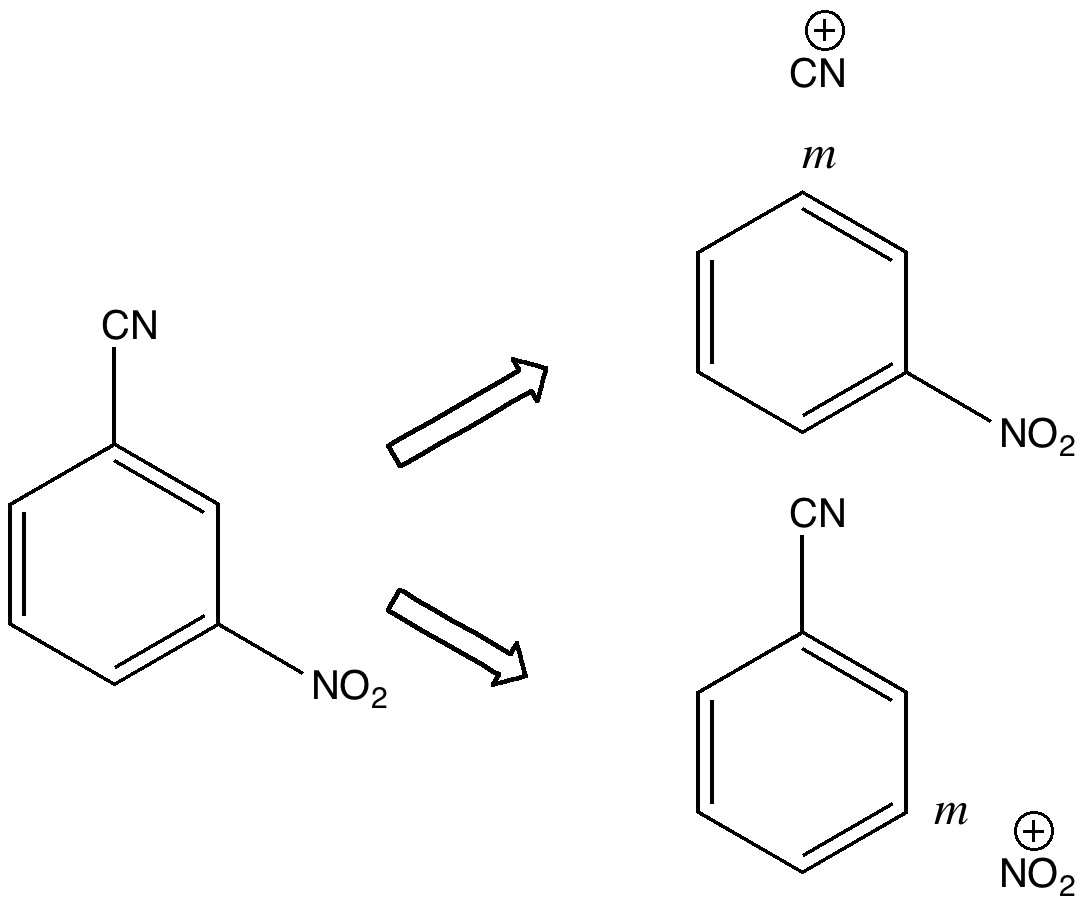
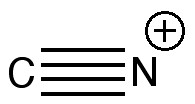
One teaches how to disconnect each group, identifying that both are meta-directing towards electrophiles, and hence asking what an appropriate electrophile might be? The “correct” answer is a nitronium cation (nitric + sulfuric acids) acting upon m-directing benzonitrile. But (sacrilege), why not a “cyonium” cation (CN+) on m-directing nitrobenzene? Well, as a tutor one would normally swat it away on the grounds it has never been previously observed (or that cyanide is always seen as an anion, not a cation). But then one (or a student) asks, why not? How about generating it from e.g. TfOCN. TfO– is a jolly good leaving group, one of the best. Well, this precursor truly appears never to have been made (or even calculated!). By now (if encountered in a tutorial), most chemistry students would be rather bemused. So the process of following one’s nose (more accurately, my nose) continues in the peace and quiet of a blog, where a rather different readership might be bemused (or inflamed).
One might start the same place a student would. How would one represent this diatomic with bonds? How about the above? It has the merit that both atoms are associated with a (shared) octet of electrons, in the form of a quadruple bond. I did show this (briefly) to a colleague, but they recoiled in horror, although it has to be said they were slightly at a loss to actually explain their horror.
Well, time for calculations. How about CCSD/aug-cc-pVTZ (DOI: 10042/to-6261) or B3LYP/aug-cc-pVTZ (DOI: 10042/to-6255). The latter allows a so-called Wiberg bond index to be computed (a reasonably accepted index). This comes out at 3.55, well on the way to being quadruple. An NBO analysis (NBO 5.9) identifies FOUR NBO orbitals with an occupancy of ~2.0, all designated BD (rather than e.g. Lp). What are these NBOs like? (as it turns out, they are almost identical to the MOs for this molecule).
Orbital 7. Click for 3D |
|
Orbital 6 (π) |
Orbital 5 (π) |
Orbital 4. Click for 3D |
|
Orbital 3. Click for 3D |
|
Orbitals 5 & 6 are standard π orbitals with no mystery (and 8 & 9, not shown, are the matching π* pair). Orbital 3 results from the overlap of two 2s AOs (but note the curious little toroid at the carbon end). Orbitals 4 and 7 (the LUMO) are the interesting ones. Nominally, the result of overlapping two 2px AOs to give what should be a bonding and antibonding pair, they both appear to be bonding in the C-N region! Perhaps the quadruple bond is not looking quite so unlikely after all (comprising ~double occupancy of orbitals 3-6)!
What about those stalwarts I often use in these blogs, QTAIM and ELF? The former (using the CCSD natural orbitals) has a ρ(r) of 0.346 and a ∇2ρ(r) of +2.01 at the bond-critical point (BCP). The former is certainly a high value, although no calibration exists to compare it to a quadruple bond. The Laplacian has a positive value at this point, possibly an indication of a charge-shift bond (see this and this blog, although more likely due to the adjacency of the bond critical point to the core shell of the carbon atom). ELF (also using natural orbitals) declares the presence of TWO disynaptic basins, with integrations of 5.39e and 2.44 (totalling 7.83e). The basins will each take the form of a torus (see DOI: 10.1021/ct100470g). Hm, perhaps, on reflection, this paragraph might not be entirely suitable for an introductory tutorial to organic chemistry. The density of mumbo-jumbo is rather high!
So starting from a simple retrosynthetic analysis of a simple aromatic molecule, in which the less obvious route is at least considered, one derives a “new” reagent, the cyonium cation CN+.In a effort to analyse its bonding, one concludes that a quadruple bond needs to be taken at least seriously. I would note as a warning that these diatomic species can be really tricky to pin down, and the iso-electronc C2 is a good example of that. But C2 has all sorts of issues, some of which are avoided with CN+. So the last word is hardly written, but not a bad outcome, I venture to suggest, of following one’s nose in a tutorial.
I have appended to this post a 3D exploration of the ELF function, showing the two torus basins referred to above.
ELF function for CN+. Click for 3D
Henry Rzepa, URL:http://www.ch.imperial.ac.uk/rzepa/blog/?p=3065. Accessed: 2011-06-04. (Archived by WebCite® at http://www.webcitation.org/5zBSjBjhM)
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
{kind=link}
View Comments
So, how would one prepare "cyonium triflate" TfOCN?
Or should it be cyanium? And perhaps Tf2NCN, or Tf3CCN ?
The electronic structure questions you ask are the same ones that many inorganic chemists have pondered in relation to what makes CO/CN- such fantastic end-on ligands. Orbitals 4 and 7 should correspond to the anti-bonding combination of the 2s orbitals and bonding combination of the px orbitals in a first-order PMO framework. Second-order mixing, or sp-mixing depending on your nomenclature, results in both orbitals being approximately non-bonding in character. The exact nature of this orbital has been debated extensively, and two papers you may find interesting and relevant are 10.1021/ja960876z and 10.1021/jp972657l.
Thanks for the two links! Yes, it is an irony that the simplest of molecules cause the greatest theoretical difficulties. C2 itself has multi-reference character, and it may be that CN+ itself also has. This presumably means that any one canonical MO must almost inevitably present a very incomplete picture.
Ahh yes, of course and your optimized singlet structure has a wavefunction instability (note: use stable(opt,xqc) as it has trouble converging without the xqc flag). Some interesting notes after running a few calculations at the same level of theory (B3LYP/aug-cc-pVTZ):
R(CN-) = 1.1714 Å
R(CN+, closed-shell singlet) = 1.1724 Å
R(CN+, open-shell singlet) = 1.2341 Å
R(CN+, triplet) = 1.2312 Å
In all of the structures considered, the bond elongates upon removal of two electrons from cyanide! It seems that removal of the electrons from the orbital that becomes your LUMO suggests it to be truly non-bonding (del-R = +0.0010 Ang), whereas a noticeable elongation (~0.05 Ang) is observed for the other two states.
At this level of theory, the triplet is predicted to be lowest in energy followed by the open-shell singlet (+6.43 kcal mol-1) and finally the closed-shell singlet (+25.98 kcal mol-1). Both the triplet and open-shell singlets can be characterized as having three electrons in the pi orbitals (its the oxidation from this orbital that seems responsible for the large structural change), and one electron in the orbital corresponding to your LUMO.
Thus, if cyonium can be synthesized, it may be spin-forbidden from participating in very likely reactions!
Thought-provoking post, but I couple of comments.
CN+ has similar multi-reference character as C2 [see JCP, 112, 9213 (2000), for example, which discusses FCI and very high-order MPn results for ground state CN+ (which is the singlet Σ state)]. I think this multireference character underlies the curious results presented above. From a simple MO picture, It's unclear to me why CN+ would be qualitatively different from C2.
Similarly, the statement that "this precursor truly appears never to have been made (or even calculated!)" seems a bit inaccurate. CN+ certainly has been studied extensively in the gas-phase for at least half a century, and plenty of calculations have been performed. However, I'm unaware of any calculations designed to specifically look at the possibility of a quadruple bond.
To clarify just one aspect, the "precursor" I was referring to was TfOCN, not CN+ itself. More specifically, CAS appeared to have no entries for this molecule, or anything particularly close.
Sorry, that makes more sense that you were speaking of TfOCN, not CN+ itself.
Regardless, this paper by Hiberty and co-workers that just came out on the bonding in C2 caught my eye and seems relevant:
10.1021/ct100577v
The article Steven refers to is a comprehensive analysis of C2 using valence bond theory, with the conclusion that the largest contributor by weight is a triple bonded formulation •C≡C• (and that the conventional picture of a suspended double bond with no underlying σ-bond plays only a marginal role). There is also an interesting discussion of the significant charge-shift character of the two π-bonds. I have discovered a plot of the Laplacian isosurface I did a little while ago which appears to show this feature (blue = charge depletion = +ve Laplacian = charge shifted).
I fancy the story will not stop here, and it will be interesting whether VB theory reveals if any quadruple bond characteristics might also be present and what their weight might be (the article does not discuss this possibility).
For good measure, here is the Laplacian isosurface for CN+.
It is both similar to C2 in the charge-shift character of the π-system, and yet significantly different.
All this discussion never mentions my seminal 1993 paper, "Is a CC Quadruple Bond Possible," Schleyer, Maskak, Chandrasekhar, Grev, Tettrahedron Letters, 34, 6387-6390, which finds 4.00 Wiberg and NPA/NLMO bond orders for N2(2+), Si2, and P2(2+) states as well, albeit for the "wrong reason," as explained in the paper.