In Internet terms, 23 years ago is verging on pre-history. Much of what was happening around 1997 on the Web was still highly experimental and so its worth taking a look at some of this to see how it has survived or whether it can be “curated” into a form that would still be useful. I had noted in my earlier comment a site which early on had become non-functional and then speculated whether any volunteers might have suggestions for how to best rescue it.
There are two ways of approaching any such rescue operation; a manual editing of the code behind the site (the HTML) or a more automated approach to doing so. The site in question in fact probably has more than 200 HTML documents that would need such an edit, which is impractical (or costly) for human curation. But the underlying well-formed structure of HTML lends itself to automation and now a saviour in the form of Ángel has indeed come forward with the solution!
One of the least stable aspects of Web pages written in the period 1993-1998 or so was the manner in which extensions to perform specialised tasks were handled. The first solution in chemistry[1] was to use the Web page itself to launch an external molecular viewer such as Rasmol via a protocol known as MIME,[2] but that depended very much on the viewer already being pre-installed on the device being used. This was a shop-stopper if you did not have the administrative rights to do so. Netscape was a company set up in 1994 whose main product was an innovative browser which could be extended by “embedding” a window directly into the display page using a plug-in rather than the earlier solution of having a separate window. In 1995, one such plugin appeared for the Netscape browser called “Chime”, which allowed 3D coordinates representing a molecule to be displayed as an interactive model within the page. The plugin still had to be pre-installed by the user and this is how in 1997 https://www.ch.ic.ac.uk/vchemlib was set up to function.
The limitations of plugin pre-installation soon became apparent. A partial solution was to download the plugin as part of invoking the web page itself. For this to work across a range of different devices running different operating systems, the plugin had to work on all of them. The solution was based on Java applets, which in turn would still rely on an initial underlying installation (with admin rights) of the JRE (Java Run-time Environment) on the device. This would now support a wide variety of different Java applets, rather than requiring each of them to be pre-installed by the viewer of the page. Between the period 1998 or so up to around 2015, the functionality of the Chime plugin was implemented and indeed greatly extended into the Java-based Jmol applet.[3] Unfortunately, using this did now require rewriting the underlying HTML code for each individual Jmol invocation.
The next step brings us up to the present method, which was to replace the Java applet by a Javascript-based module which would NOT require a JRE to be pre-installed. All the required installation would be handled by the browser itself; the runtime environment in effect was now built into browser itself. This again required a change to the HTML code for the invoking this tool. So the nature of the curation required to revitalise https://www.ch.ic.ac.uk/vchemlib/ can now be defined: replace the HTML code used to invoke Chime by new code which invokes its current replacement, JSmol (which stands for JavaScript Jmol‡). The good news is that this is a simple programmatic procedure, which itself can be implemented using Javascript. Here is where Angel comes in. He has freshly written convert.js as a script which performs this task. It is now invoked by simply adding a header to every HTML document as <script src="convert.js" type="text/javascript"></script> and all the necessary conversion from the old Chime syntax is then done on the fly when the page is loaded.
The big win is that as a toolkit, JSmol is very much more capable than Chime ever was! One of the many interesting things it can do that was not previously possible is “computation”. I thought I would illustrate how this veritable resource has not only been curated back into (mostly) working order, but also how its functionality as a molecular toolkit has been greatly enhanced.
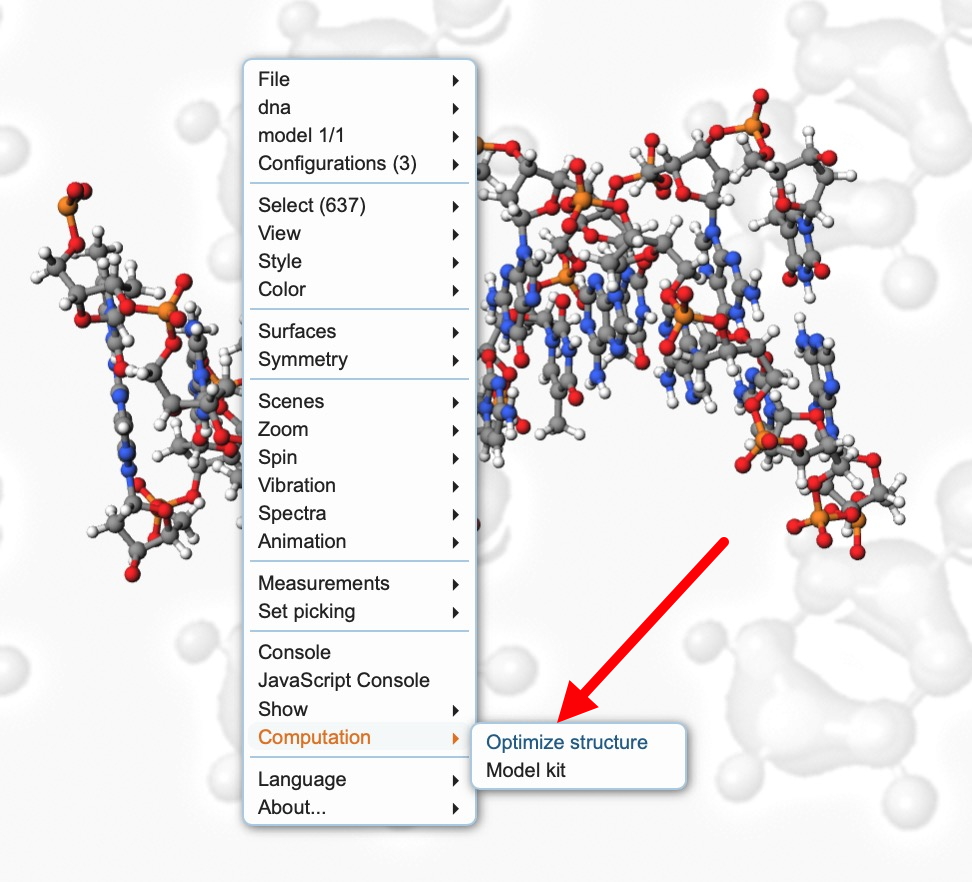
We are going to illustrate this using the tool optimize structure, the menu for which can be invoked by a right-mouse click anywhere in the molecule window. What does this mean? Well, I need to start by covering the basic sources for 3D molecular coordinates, which can be generated using a wide variety of methods, some of which are listed below.
- They may be derived from simple 2D flat diagrams such as produced by e.g. Chemdraw, with some indication of 3D using chemical hashes and wedges. The “z” coordinate can be zero for all atoms. Clearly not optimal.
- A 3D structure can be generated from a 2D one using very simple rules about the 3D environment about each atom, such as tetrahedral carbons and simple standard bond lengths and angles. Programs such as Avogadro[4] can do this as part of loading a molecule with only 2D coordinates present.
- This simple rule can be extended to using a full force field, which includes much more information about bond distances, bond angles, torsions, inclusion of van der Waals attractions and repulsions and electrostatic effects (but significantly not any effects based on electronic structure).
- Full-blown quantum mechanical computation of the geometry, including electronic effects.
- Experimental coordinates such as obtained using crystallography.
In general, information on which of the above categories were used to obtain the 3D coordinates are infrequently, if ever, actually declared on the web page. Indeed, this information for the site https://www.ch.ic.ac.uk/vchemlib/ is missing, only the original author might know! Of these types, #3 is computationally fast enough to be implemented into a Javascript such as JSmol, so we can now test how “optimised” any set of 3D coordinates actually is (#4 is not yet possible). Here are some instructions on how to proceed. For illustration I will use this molecule from the site, accessed as https://www.ch.ic.ac.uk/vchemlib/mol/direct_pdb.html?senses/vision/colour/pdbs/carotene.pdb The coordinates are expressed in the so-called PDB format, which was originally developed with proteins in mind and not small molecules.
- Load the link above and right-click to bring up the toolbar menu shown below:
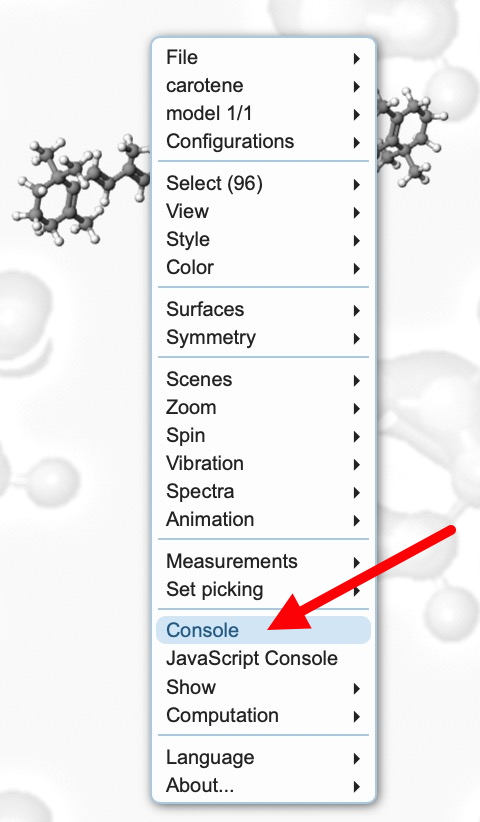
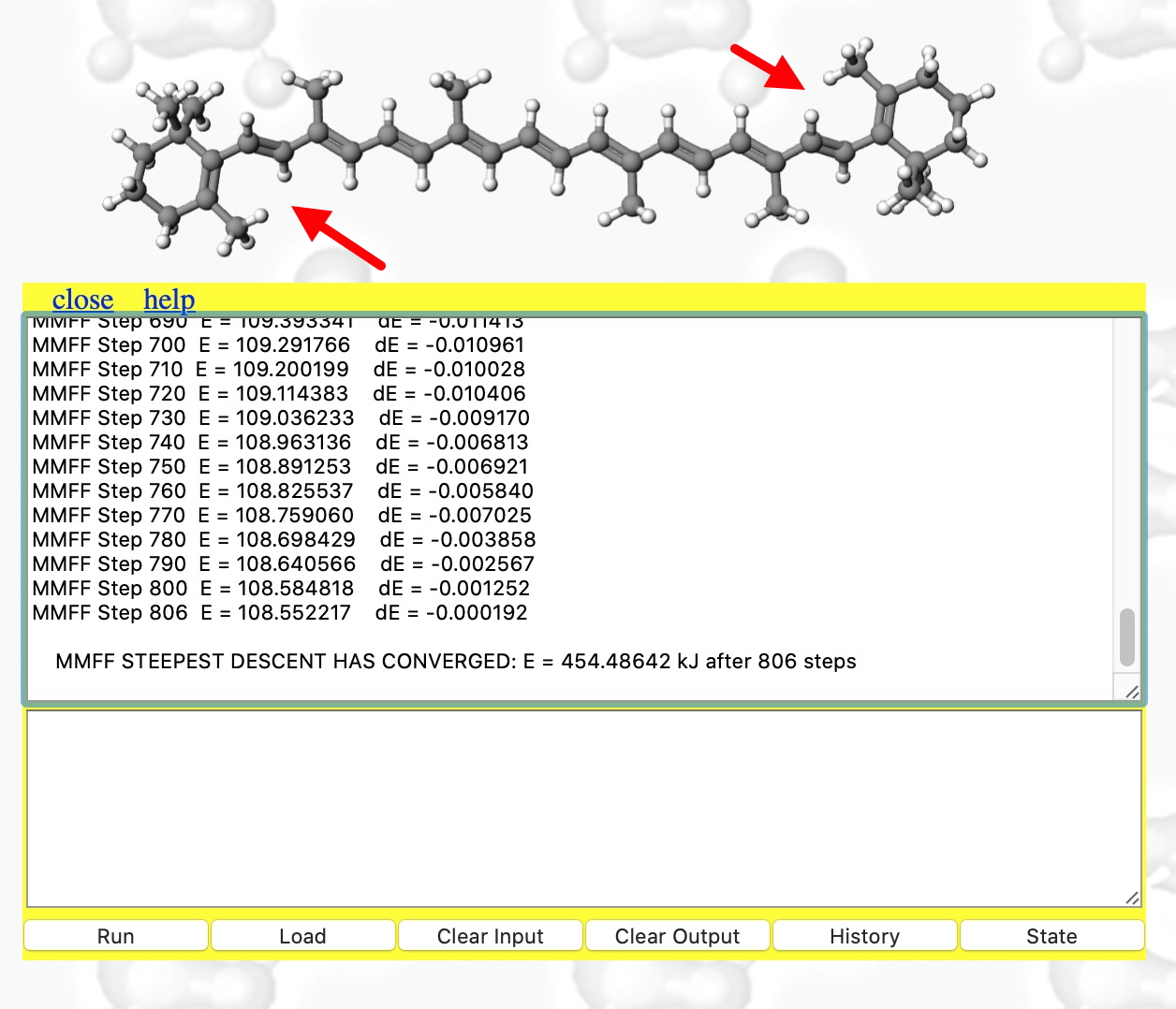
- When doing any computation (especially one that might turn out to be slow!), it is useful to get feedback and this is done by opening the Console. With the molecule now available to inspect, you might notice some anomalies♠ indicated with red arrows.
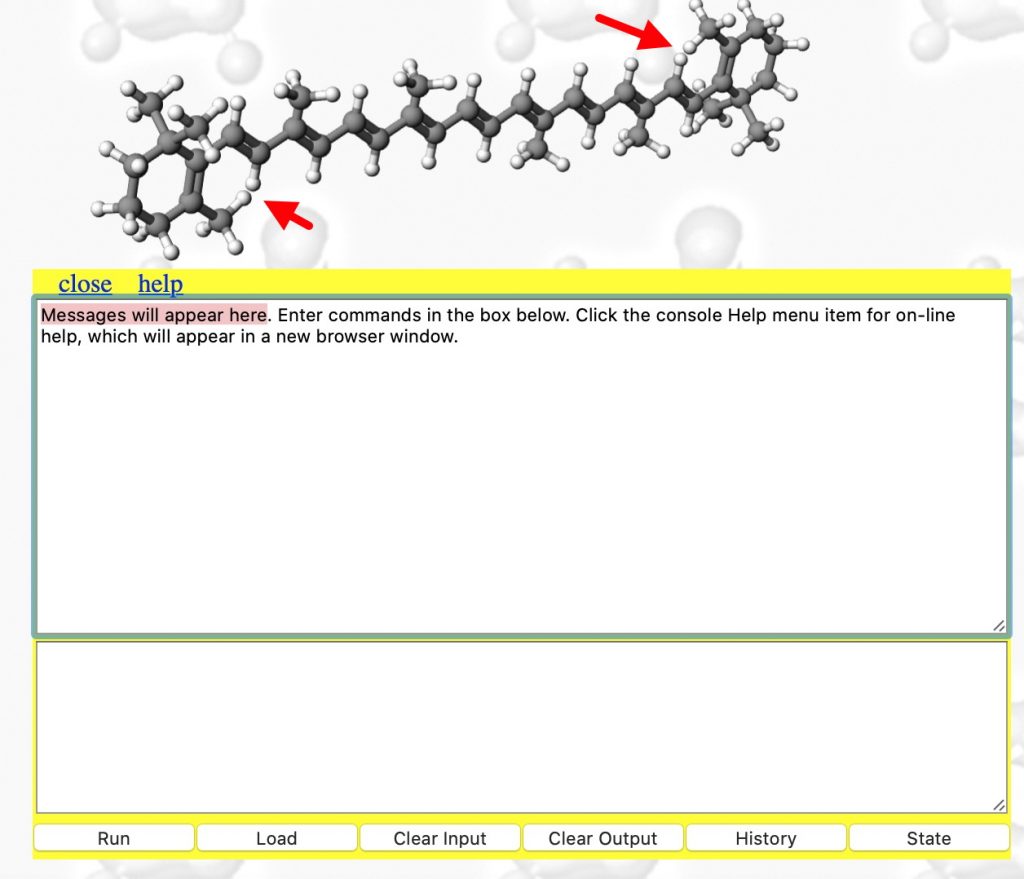
- The top panel of the Console shows JSmol responses and the bottom panel is where you can type commands for JSmol. Type the following commands one at a time into this bottom panel, each ending with pressing the return key:
- set forcefield "MMFF94”
- set minimizationMaxAtoms 400
- minimize steps 100
- set forcefield "MMFF94”
- This produces the result shown below. The MMFF94 force field has been selected, the maximum atom count set to 400 (default is 200) and 100 steps of energy minimisation requested (the default). The energy E is the so-called steric energy, which is the sum of all the terms given in #3 above. The fact that it starts with a value of 27606 kJ/mol† and reaches 157 kcal/mol after 100 iterations suggests that the 3D coordinates were indeed far from optimum; E is normally in the range -300 to + 300 kcal. Notice also the dE is the change in energy every 10 iterations. You really need to get this down a bit lower, so repeat the minimise instruction (and set the max steps to a larger value such as 1000)
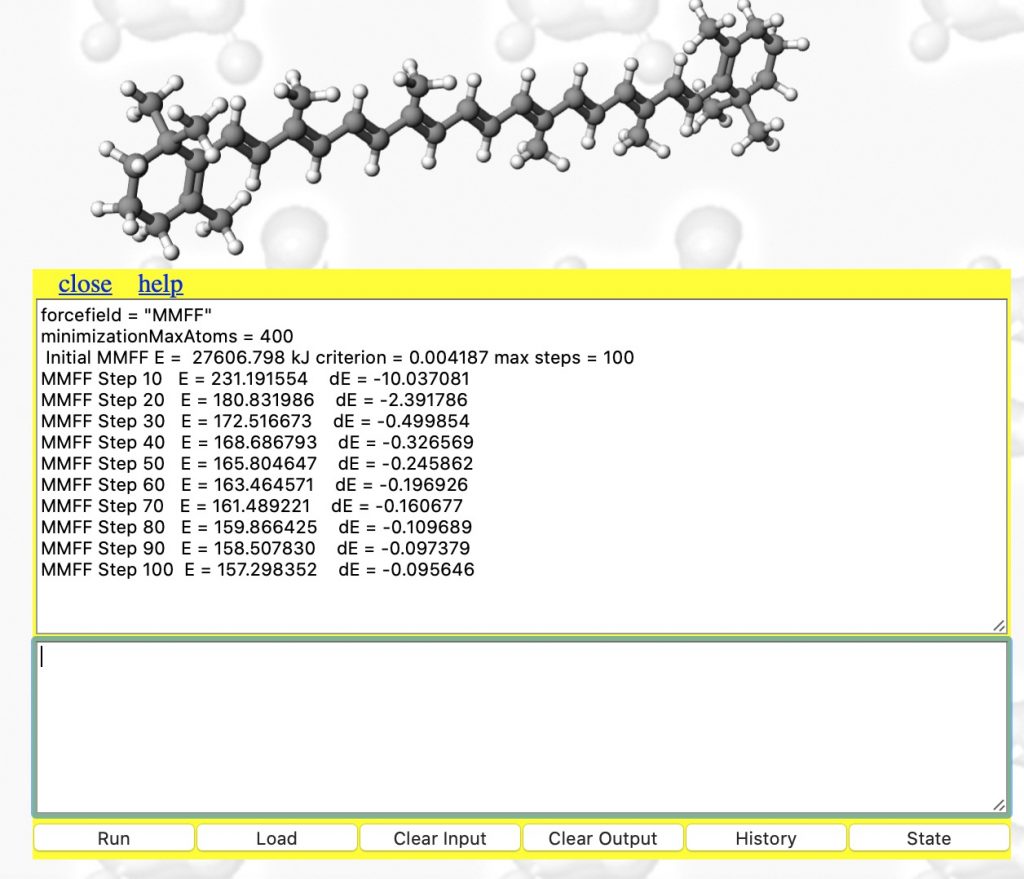
- The minimisation finally converges after 806 cycles (a default of 100 is rarely enough) to 108.6 kcal. To update to the final geometry, enter a return in the bottom panel. Inspect the region indicated with red arrows again!
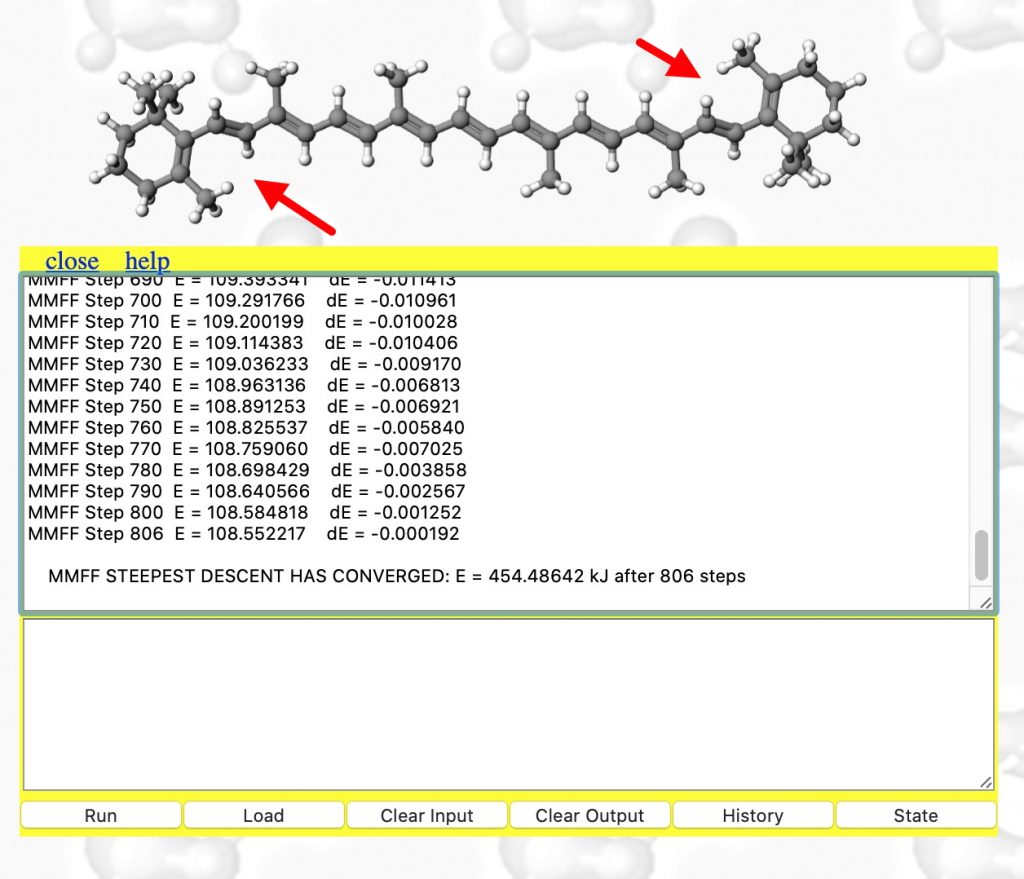
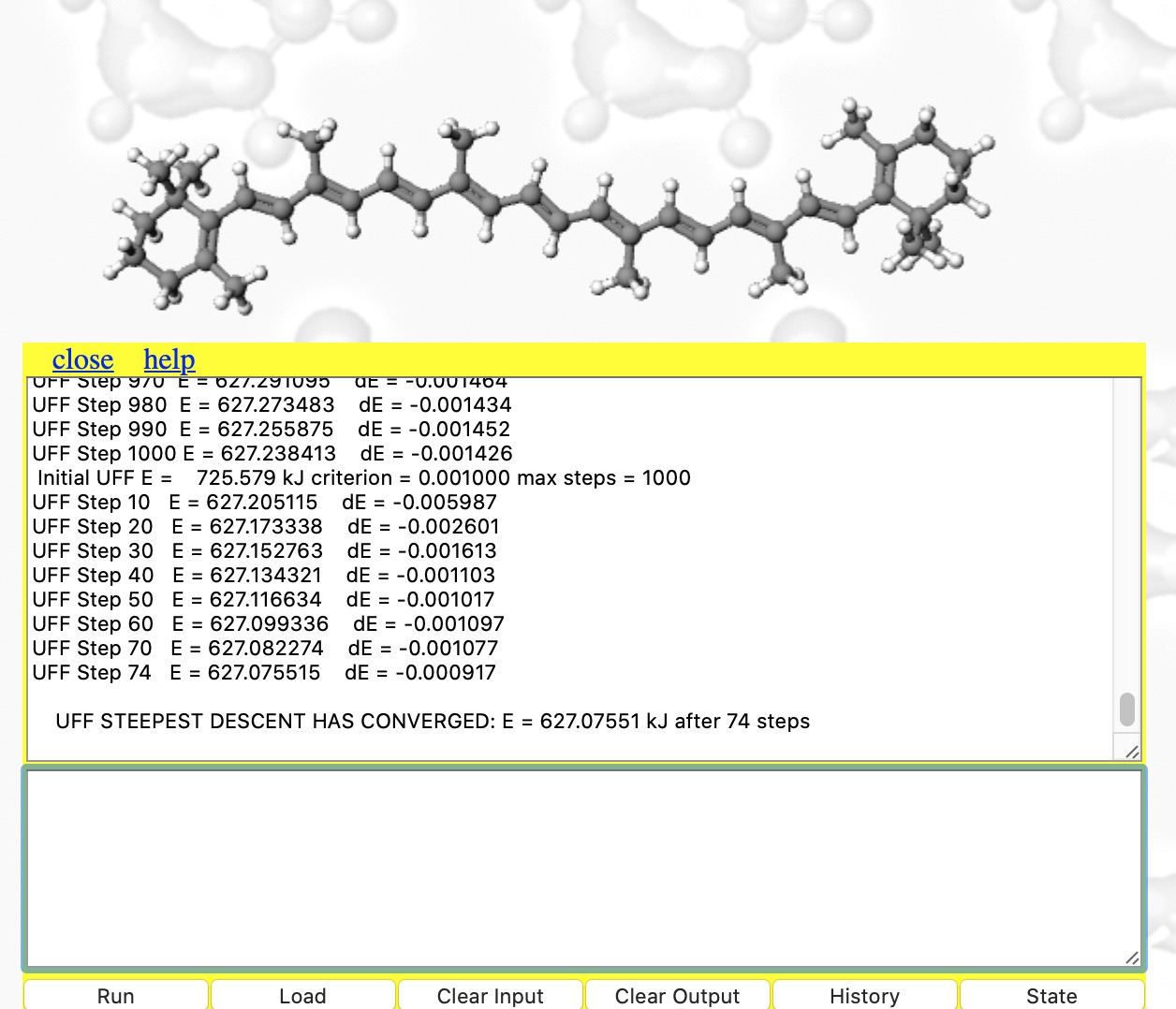
- Now type set forcefield "UFF" into the bottom panel and repeat the minimisations until convergence is obtained (about 4000 cycles!). This is using a much more approximate force field, but one that is applicable to most elements in the periodic table. The initial UFF energy is 1086 kJ/mol and the final one 627, a much smaller change than before (absolute MMFF94 and UFF energies themselves cannot be compared) accompanied by only a small change to the final geometry.
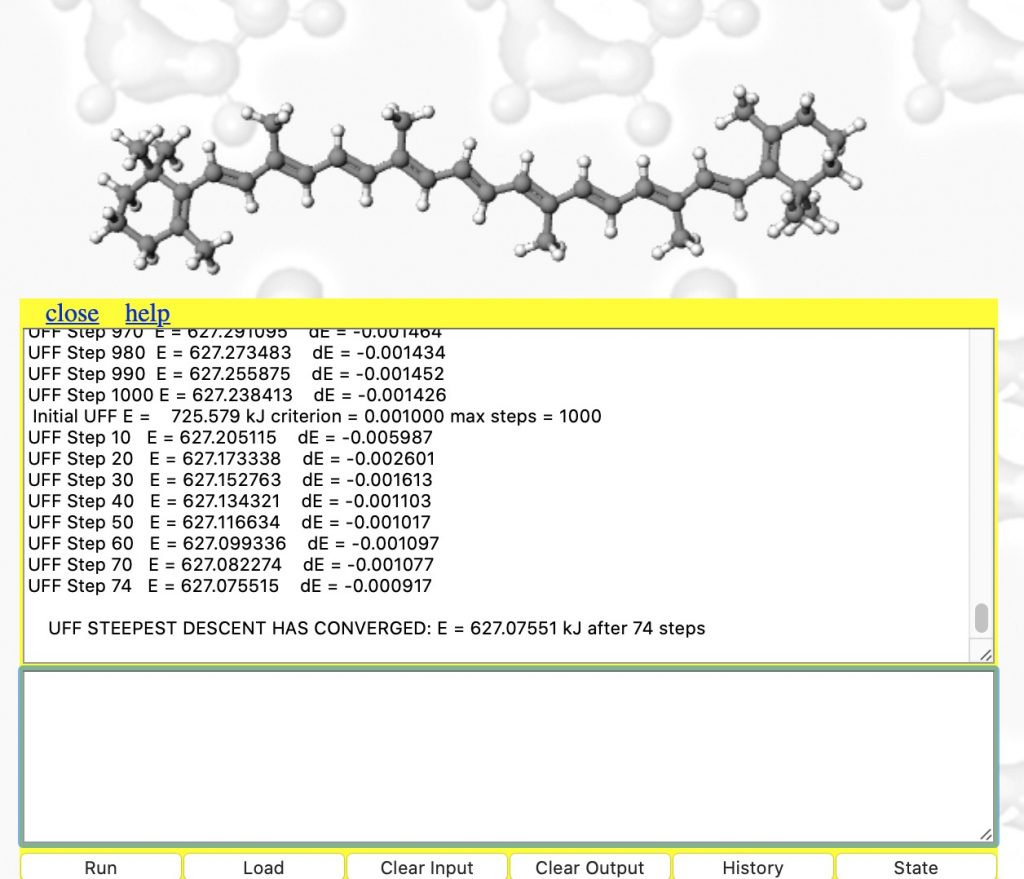
- Now type e.g. write beta-carotene.pdb into the bottom panel to download the final and now optimised geometry file to your device.♥ You might as well put all that hard-earned optimisation to good use elsewhere.
- I will end with an experiment to highlight an issue intrinsic to force field optimisations. The force field operates by identifying standard environments for the atoms and bonds in the molecule, such as the atom hybridisations and assigning the correct type of force constant to them. If the molecule has not been defined correctly, this process cannot be done. In these instances, only the UFF field can be used. Then try this example:
https://www.ch.ic.ac.uk/vchemlib/mol/direct_pdb.html?polymer/synth/acrylates/pdbs/methyl_methacrylate.pdb
and try to select the MMFF84 force field and mimimize. It will instead use the UFF field, almost certainly because the so called CONECT records in the PDB file are incomplete or incorrect. Nowadays, PDB is rarely used for these sorts of purposes, with e.g. a Molfile or CML format being preferred. This has much more reliable connectivity and bond type information baked into it. This sort of issue can be a real problem for larger molecules, since there are 100s of connection records defined and even a single error in any of them can prevent a good force field from being used. Even an experimentally derived set of coordinates such as from a crystal structure will still require atom and bond types to be correctly assigned. The general solution to this sort of issue is to move over to a quantum mechanical (QM) treatment, where atom and bond types are not used at all. Instead the only information needed is the atom list and a set of approximate coordinates (and charge if the molecule is not neutral together with spin state). Unfortunately, implementing a QM procedure into JSmol would require computers that are perhaps a factor of ten faster interactively than current ones. Not impossible to envisage and perhaps the next improvement to this site in another 10 years time!
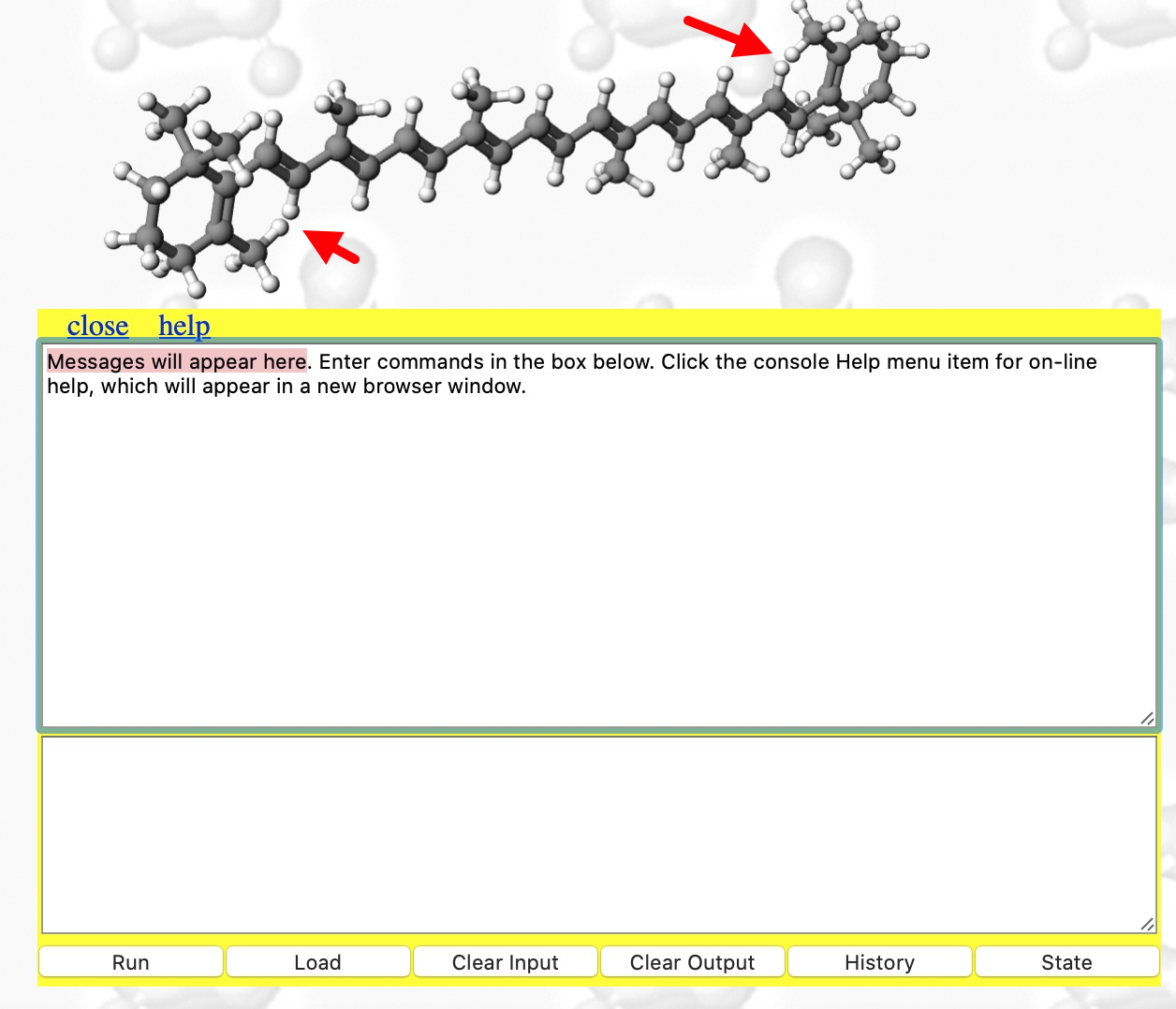
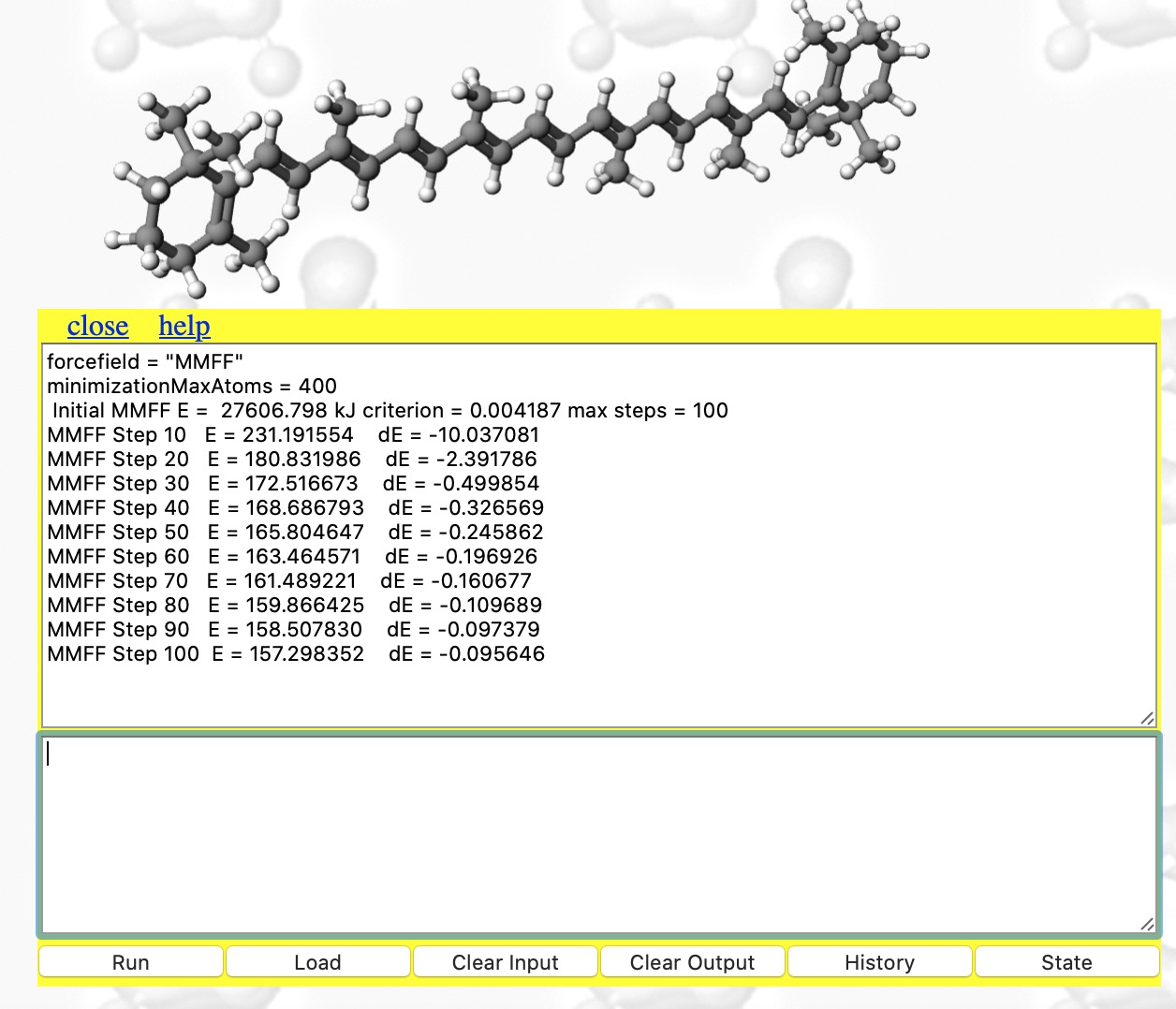
The concept that a Web-based resource like this can provide a chemical toolkit embedded within the page to conduct experiments such as the ones described above was nonetheless very much the original intention envisaged all those years ago.[1]
‡Just to clear this up, Java and Javascript are NOT the same despite the name. †This is implied as kJ in this version of JSmol. ♥You might as well write out carotene.mol or carotene.cml, which are better suited for further processing with more reliable bond records. The latter was indeed designed to avoid any loss of information during such conversions if at all possible! ♠A similar anomaly formed the basis of this critique of the vibrational mode imaging of a tetraphenylporphrin.
Author
References
- O. Casher, G.K. Chandramohan, M.J. Hargreaves, C. Leach, P. Murray-Rust, H.S. Rzepa, R. Sayle, and B.J. Whitaker, "Hyperactive molecules and the World-Wide-Web information system", Journal of the Chemical Society, Perkin Transactions 2, pp. 7, 1995. https://doi.org/10.1039/p29950000007
- H.S. Rzepa, P. Murray-Rust, and B.J. Whitaker, "The Application of Chemical Multipurpose Internet Mail Extensions (Chemical MIME) Internet Standards to Electronic Mail and World Wide Web Information Exchange", Journal of Chemical Information and Computer Sciences, vol. 38, pp. 976-982, 1998. https://doi.org/10.1021/ci9803233
- R.M. Hanson, "<i>Jmol</i>– a paradigm shift in crystallographic visualization", Journal of Applied Crystallography, vol. 43, pp. 1250-1260, 2010. https://doi.org/10.1107/s0021889810030256
- M.D. Hanwell, D.E. Curtis, D.C. Lonie, T. Vandermeersch, E. Zurek, and G.R. Hutchison, "Avogadro: an advanced semantic chemical editor, visualization, and analysis platform", Journal of Cheminformatics, vol. 4, 2012. https://doi.org/10.1186/1758-2946-4-17
Very nice piece, Henry!
I’m so glad to have rescued this website. The collection of molecules is remarkable, even if not all the structures are very refined in terms of geometry.
It’s a shame the large amount of educational websites that have been lost due to the technical issues, associated to browsers dropping the support first for plugins and later for Java applets. Fortunately with some effort and time they can be brought to life as long as the original source files are available, as it was in this case. This is all thanks to the Jmol project, which enjoys a continuous development effort (mainly Prof. Bob Hanson) and a lively community of users and authors of materials, both in education and research.
Thanks indeed Angel.
Yes, the genius in the creation of the Web, back in 1989, was to make “source code” an intrinsic part of the design. The ability to “reverse engineer” the early sites is how many people learnt how to write new pages in the first place. I remember about ten years later commercial companies (no names here but you can probably guess who they were) tried to convert the Web to binary (proprietary) code. Thankfully that never took off!
I will tell a story about a product I think was called LabSkills. Our department purchased it perhaps 15 years ago, and it was not inexpensive. It was written in Adobe Flash. Importantly, the source code was not accessible. A few years later we wanted to add some content, but were told that would not be possible, we would have to take out a new license to gain content. Now the Flash player is ceasing in a month or so, and browser support for it will vanish. No chance however of rescuing the Labskiils content in the way that the Vchemlib site has been.
The conversion library is now available at doi: 10.5281/zenodo.4252726