In 2013, I created an iTunesU library of 115 mechanistic types in organic and organometallic chemistry, illustrated using video animations of the intrinsic reaction coordinate (IRC) computed using a high level quantum mechanical procedure. Many of those examples first derived from posts here. That collection is still available and is viewable in the iTunesU app on an iPhone or an iPad. The realisation struck me now‡ that one of the types not described in that library was Michael-type 1,4-nucleophilic addition to an activated alkene, as described at Wikipedia. So here is that addition.
The base used will be NH3 and the activating groups R will all be formyl. The DFT computational method will be ωB97XD/Def2-TZVPP/SCRF=water and the FAIR data will collect at DOI: 10.14469/hpc/7027
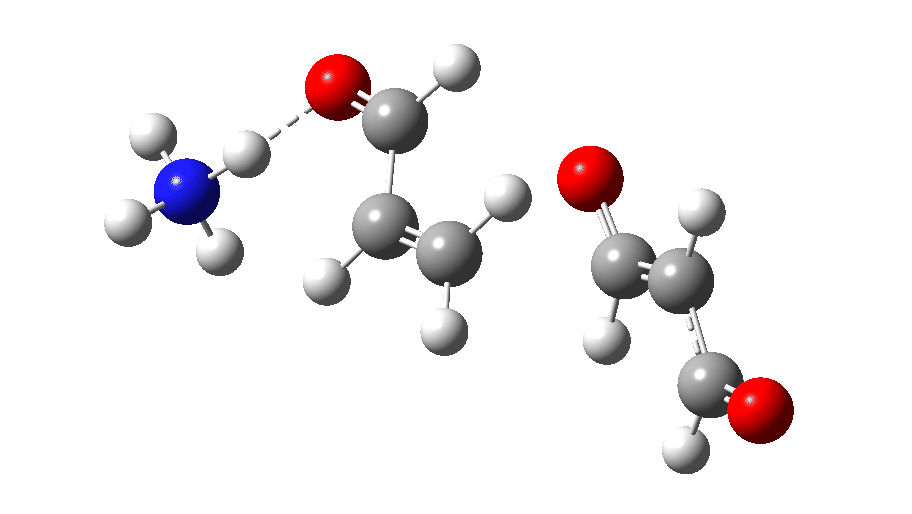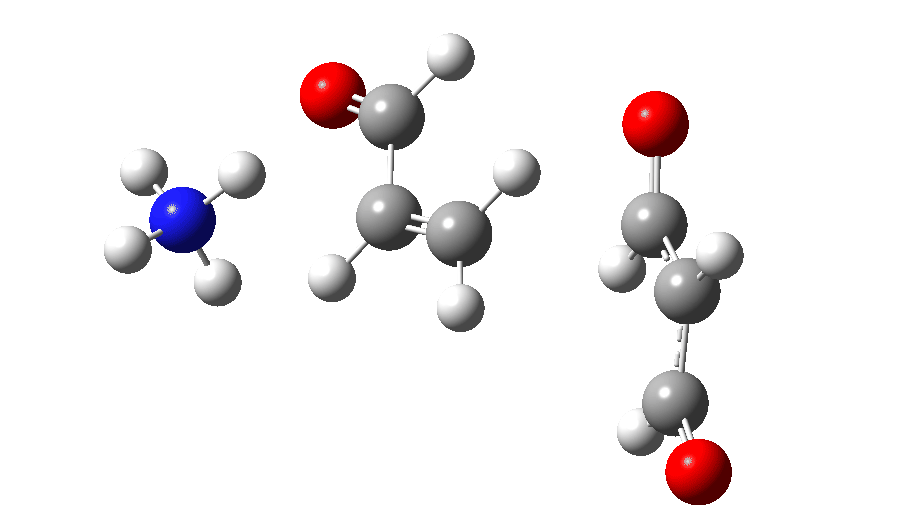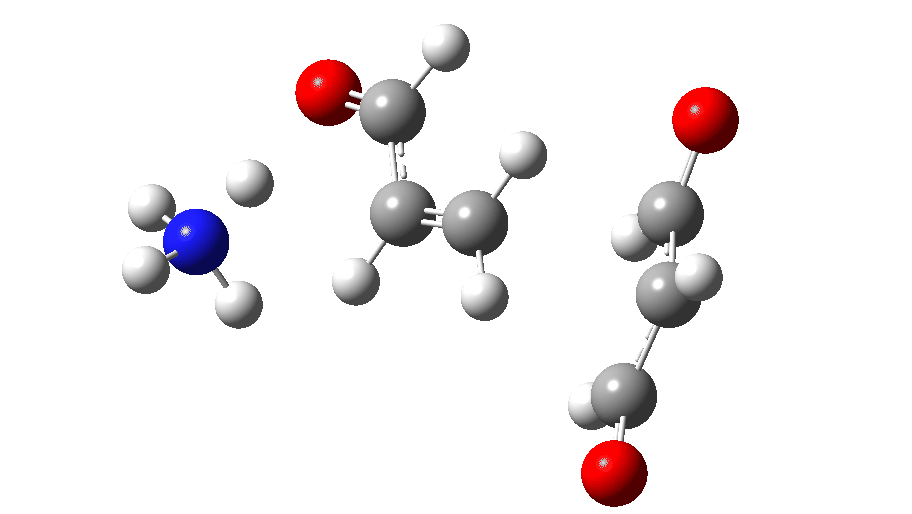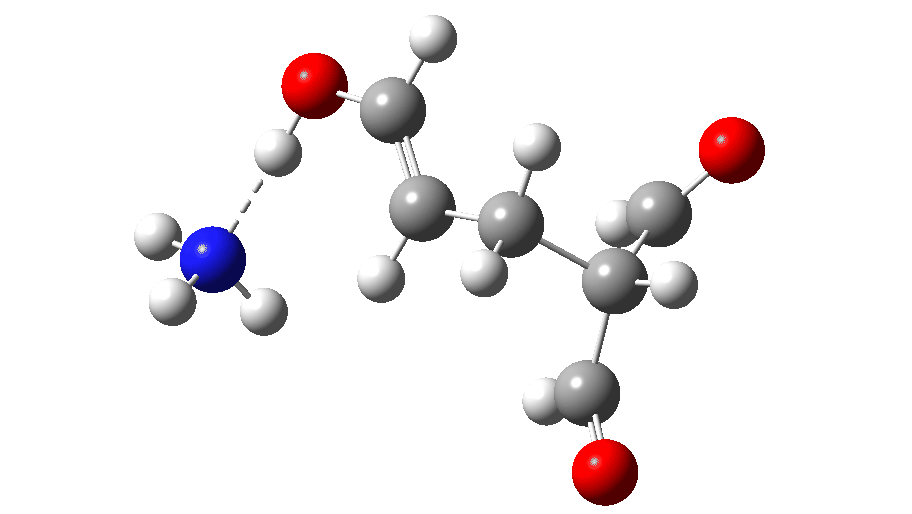
The full reaction mechanism can be represented as below
| Species | ΔΔG298, kcal/mol | FAIR Data DOI |
|---|---|---|
| Reactant | 0.0 | 7028 |
| TS1 | 6.7 | 7029 |
| Int1 | -7.7 | 7036 |
| TS2 | 16.3 | 7031 |
| Int2 | -8.7 | 7033 |
| Int3 | -8.7 | 7035 |
| TS3 | 9.6 | 7030 |
| Product | -13.2 | 7034 |
The rate-limiting step of C-C bond formation is coupled with almost synchronous protonation on the remote oxygen. It is driven by reducing the dipole moment of the zwitterion Int1, as shown below.
Attempts to find an analogous route with carbon protonation leading directly to the product did not succeed.
By varying parameters such as the nature of the R groups or the base, one might be able to control the choreography of the C-C bond formation relative to the accompanying proton transfer to oxygen in TS2 (in the manner that was possible for e.g. peracid epoxidation[1]). These changes could then be subjected to e.g. the measurement of kinetic isotope effects and comparison with values calculated from the computational mechanism.
‡With Coronavirus now changing our lives and our work patterns, and having done my allowed quota of one exercise walk for the day at 06.30 (to avoid social contact, although in fact the park we went to had lots of other people exercising, even at that time) I settled down to think about what else could be done. The Michael reaction suddenly appeared! Locating transition states is one of those things that gives me considerable pleasure, and I have not reported any for a few posts now.
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
{kind=link}
{kind=link}
{kind=link}