Pursuing the topic of halogen bonds, the system DABCO (a tertiary dibase) and iodine form an intriguing complex. Here I explore some unusual features of the structure HEKZOO[1] as published in 2012[2] and ask whether the bonding between the donor (N) and the acceptor (I-I) really is best described as a “non-covalent-interaction” (NCI) or not.
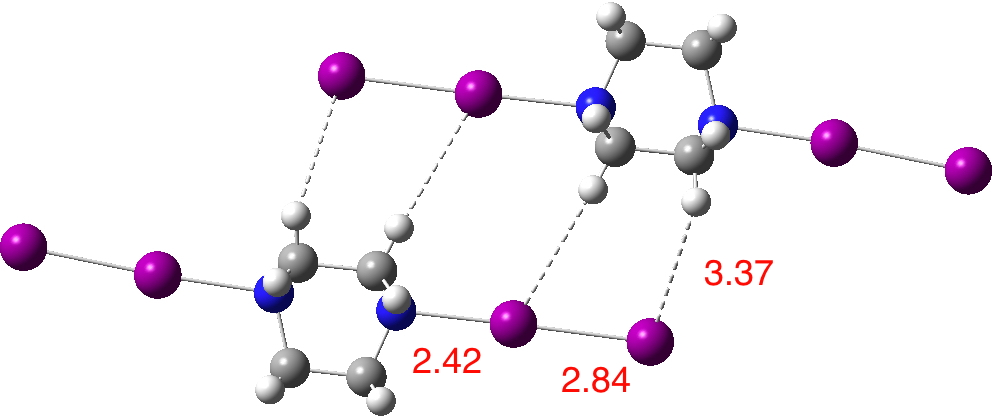
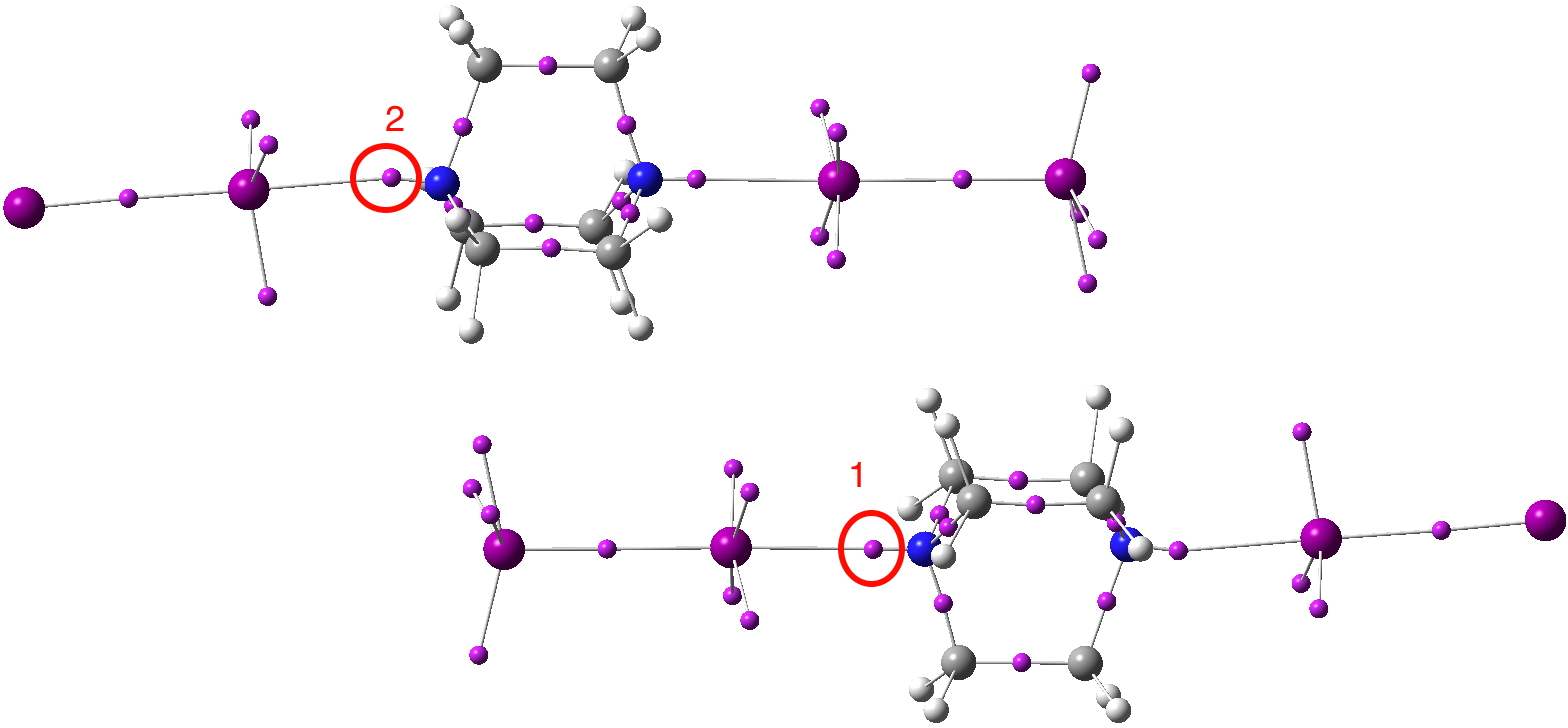
The crystal structure shows a repeating unit, with each DABCO surrounded by two I2 molecules aligned along the N-N axis. These linear chains are stacked in a dislocated manner induced by aligning to optimize the dispersion interactions between the iodines and the CH groups. The most surprising aspect is the N…I distance shown as 2.42Å. The van der Waals radii of N and I are respectively 1.55 + 1.98 = 3.53Å, a contraction of 1.1Å. The covalent radii are 0.75 + 1.33 = 2.08, an elongation of 0.34Å So is this a strong (non-covalent) interaction/contraction or a stretched weak covalent bond?
I will start with a B3LYP+D3/Def2-TZVPP+PP calculation on the crystal geometry (below).
Click for 3D
The short N…I distance has a computed Wiberg bond order of 0.26 (the I-I is 0.76) and an E(2) NBO donor-acceptor interaction energy of 40.1 kcal/mol. For comparison, the E(2) energies for conventional OH…O hydrogen bonds are around 10 kcal/mol. This makes the halogen bond a pretty strong interaction! But is it strong enough to call a bond? Indeed, do we have any means of deciding where the transition from a strong interaction to a weak bond occurs?
A NCI analysis is shown below. This analysis filters out electron densities above 0.05au (which are considered as covalent values) and shows only the properties of the reduced density gradients below this value. The dispersion attractions between the DABCO hydrocarbon chains and the iodine molecules are very apparent, but the N…I is encircled with a strange feature, normally only seen for covalent bonds or transition state breaking/forming bonds. A strong hydrogen bond for example would show up as blue, and not this strange torus.
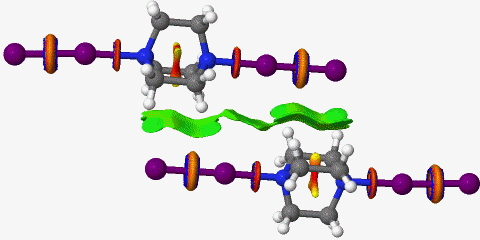
Click for 3D
Reducing the threshold for covalent densities to be filtered out to 0.03au eliminates the N…I feature (it is now covalent by definition so to speak), but the I-I feature remains. One can see here that this NCI analysis does rather arbitrarily depend on what one considers the covalent density threshold to be, and as one moves up the periodic table, this density changes. What should the density for eg a N…I interaction/bond actually be? The value appropriate for N (0.05) or I (< 0.03)?
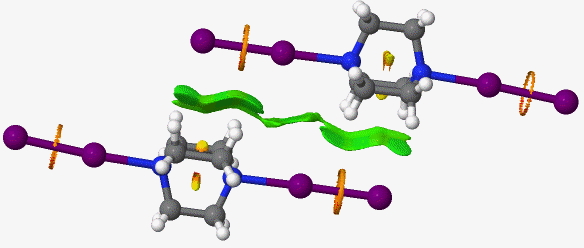
Click for 3D
Next, I show the optimised geometry of the system above.[3] This is not a periodic boundary calculation and this lack of a surrounding environment is highlighted by the change in the structure. The N…I distance lengthens, and the iodines which have no DABCO to interact with lengthen more. One might conclude that the short N…I distance measured in the solid state structure is due to some degree of N…I compression induced by the linear chains of I-I…DABCO…I-I…DABCO…I-I aligning to maximise the interchain dispersion attractions. Thus the N…I “bond/interaction” is really a co-operative effect between the N…I atoms and the extended 3D structure of the molecule. The N…I contraction of 1.1Å noted above is in part due to the intrinsic halogen bond, but also in part due to dispersion attractions elsewhere!
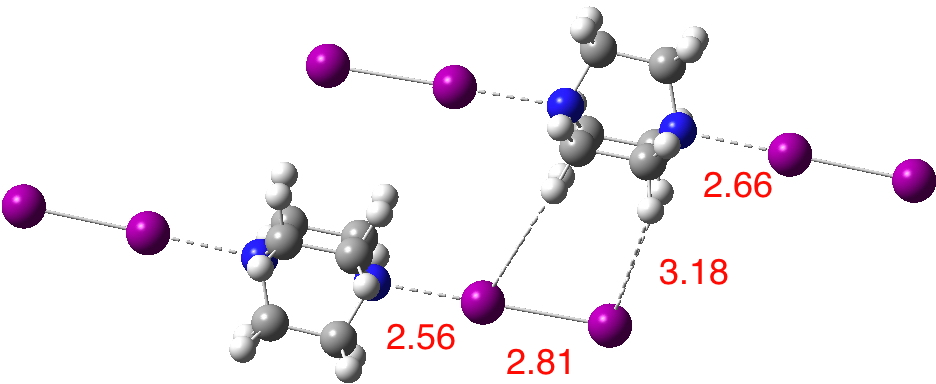
So it does rather seem as if the DABCO-I2 complex sits very much in that awkward region in which the contracted N…I distance could either be described as a weak bond or a strong interaction.‡. Just like the infamous H…H bond in cis-butene, one cannot just regard a bond as a purely localised phenomenon, one must also take into account what is happening elsewhere in the total system. Halogen bonds could also be regarding as filling the gap spanned by recognised covalency and recognised hydrogen bonds. I note in passing that another “awkward” bond is the 4th in the diatomic C2[4] that has a bond energy of about 17 kcal/mol, again weak for a normal bond and strong for an interaction.
In my next post on this theme, I will deal with another halogen bond that is found in a famous molecule known for a long time and which has another weird property.
‡Postscript. An ELF analysis reveals the following basin centroids. Basin 1 integrates to 2.00 electrons, basin 2 to 1.92. The asymmetry in the position of the basin centroid towards the nitrogen suggests it is not an equally shared covalent bond, as indeed the Wiberg bond order noted above also indicates. The I…I basin integrates to 1.86 electrons, indicating slightly reduced bonding (by donation into the sigma; orbital).
Author
References
- Peuronen, A.., Valkonen, A.., Kortelainen, M.., Rissanen, K.., and Lahtinen, M.., "CCDC 879935: Experimental Crystal Structure Determination", 2013. https://doi.org/10.5517/ccyjn03
- A. Peuronen, A. Valkonen, M. Kortelainen, K. Rissanen, and M. Lahtinen, "Halogen Bonding-Based “Catch and Release”: Reversible Solid-State Entrapment of Elemental Iodine with Monoalkylated DABCO Salts", Crystal Growth & Design, vol. 12, pp. 4157-4169, 2012. https://doi.org/10.1021/cg300669t
- H.S. Rzepa, "Gaussian Job Archive for C12H24I8N4", 2014. https://doi.org/10.6084/m9.figshare.1254779
- D. Danovich, P.C. Hiberty, W. Wu, H.S. Rzepa, and S. Shaik, "The Nature of the Fourth Bond in the Ground State of C<sub>2</sub>: The Quadruple Bond Conundrum", Chemistry – A European Journal, vol. 20, pp. 6220-6232, 2014. https://doi.org/10.1002/chem.201400356
Tags: bond energy, co-operative, donor-acceptor interaction energy
Hi Henry, You might find this one of interest!
DOI: 10.1021/jp0724525.
Another example of a halogen bond is to be found in the bromine-dioxane complex. If my memory serves, it has a very similar structure to the DABCO-iodine complex.
Nick: Indeed so, it was probably the structure that set the field on its way. It is one of the hits noted in my post http://www.ch.imperial.ac.uk/rzepa/blog/?p=13083 for D=oxygen. There are surprisingly few examples though. Thus
Sulfur is rather more common than oxygen:
Thanks Dean!
I also had a go at freezing a bullvalene (http://www.ch.imperial.ac.uk/rzepa/blog/?p=7678 and http://www.ch.imperial.ac.uk/rzepa/blog/?p=7721, but you were way ahead of me in 2007 with your approach.
I noted in the post that the “halogen bond” N…I, seemed very soft, its calculated length depending on how the lattice was constructed. Here is a rather larger lattice (DOI: 10.14469/ch/129986) The shortest N…I interactions are now predicted at around 2.45Å, fairly close to the crystallographic distance of ~2.42Å. These occur for chains where the I-I component is attracted to two separate DABCO units by dispersion forces, resulting in greater compression of the N…I halogen bond than those units interacting with only one DABCO. Shorn of any DABCO compressions, the N…I bond can extend to 2.7Å or greater. The N…I Wiberg bond index becomes 0.31 and the adjacent I…I is 0.70.