A game one can play with pericyclic reactions is to ask students to identify what type a given example is. So take for example the reaction below.

The alternatives are:
- A cyclo-elimination reaction (red arrows).
- Two concurrent electrocyclic ring openings (blue and magenta arrows)
- Two consecutive electrocyclic ring openings
- Or could it be a hybrid with characteristics of both the first two?
All the first three are four electron thermal processes; all should occur with involvement of one antarafacial mode (a Möbius transition state). But where are those antarafacial modes? Or do all or any of these pericyclic reactions not follow the standard rules? And the rules have nothing to say about whether two separate processes can be concurrent or must be consecutive.
The solution is to concede the limitations of simple electron counting rules, and evaluate the reaction using a quantum mechanical method (ωB97XD/6-311G(d,p))
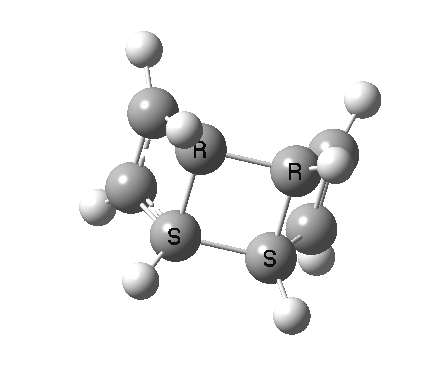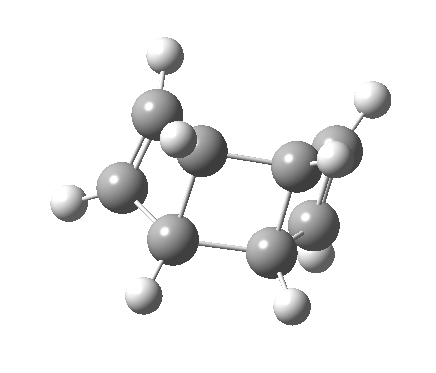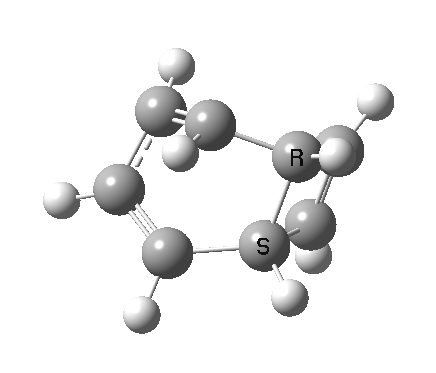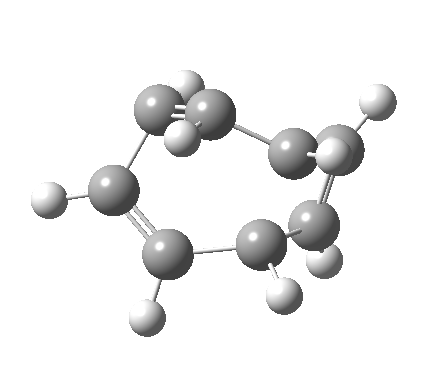
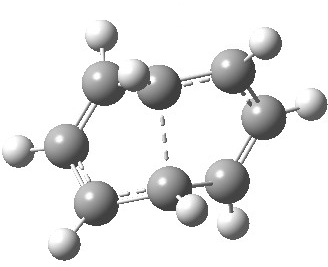
- The first attempt is to locate a stationary point with symmetry, C2 in this case. Here it is.[1]
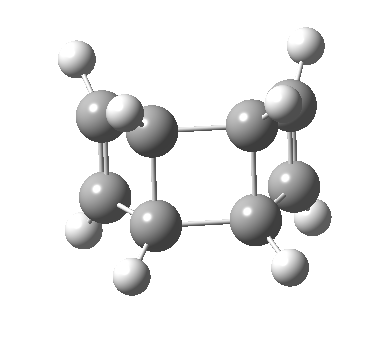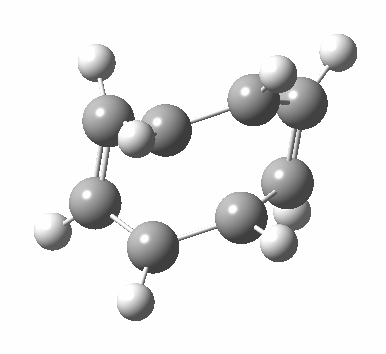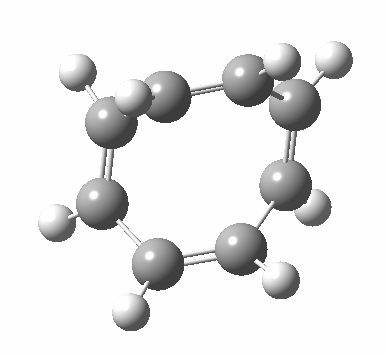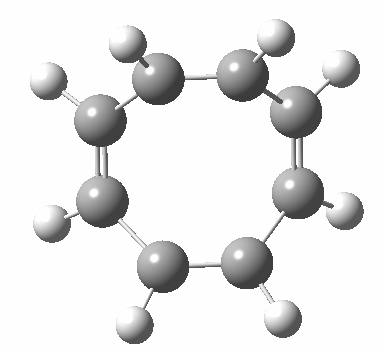

- Points of interest include the formation of the cyclo-octatetraene in the valence bond form B rather than A. This points to it being a cyclo-elimination rather than an electrocyclic reaction. For this product, little change occurs for the terminal alkenes (blue or magenta in the reactant), and indeed the C-C length at the stationary point is 1.349Å, only slightly longer than a normal alkene, and not at all the value of ~1.40Å expected for an aromatic transition state expected of an electrocyclic.
- The stereochemistry of this elimination is entirely suprafacial, as evidenced by the formation of cis-alkenes. The formal pericyclic rule that disallows such stereochemistry for a 4n-electron thermal reaction is however over-turned by the transition state adopting a trapezoidal character.
- The initial sense of rotation of the two pairs of hydrogens show above is however conrotatory (a consequence of the trapezoidal motions), as in fact required of a thermal 4n-electron electrocyclic reaction. It is only well after the transition state is passed that the motion of one pair of these hydrogens reverses itself, and we end up with a cis-alkene after all.
- So at the transition state, we see features of both a cycloelimination (trapezoidal geometry) but ALSO of two concurrent electrocyclic ring openings (conrotation). In other words, one gets the characteristics of three pericyclic reactions in one! Very much a chimera!
- There is only one fly in the ointment. This stationary point is in fact a second-order saddle-point and not a transition state.[2] There are two imaginary frequencies, and the smaller of the two corresponds to desymmetrising the two C-C breaking central bonds
- So we turn to the proper transition states in this reaction, the first of which corresponds to the initial of two consecutive electrocyclic ring openings.[3]
- This looks very different from the preceding pathway. As the (blue) arrows take effect, an antarafacial mode takes hold, ending in the formation of an intermediate bicyclic system with a trans-alkene forming. This proper transition state is 8.1 kcal/mol lower in free energy than the earlier second-order point. The C=C bond length at the transition state becomes 1.392Å, now a typical aromatic value.
- This transition state leads to an intermediate which is 12.7 lower than the preceding transition state, and is then followed by …

- a second transition state, with an energy 2.5 kcal/mol higher than the first (but still 5.6 kcal/mol lower than the second-order saddle).‡ The IRC for this step (below) in effect opens up the second ring in a follow-up electrocyclic. The C=C bond in the second cyclobutene now becomes 1.338Å, which is not characteristic of a cyclobutene ring-opening. Notice how again the initial motion of the two hydrogens of the second ring tentatively try a conrotatory motion as before, but this antarafacial motion in fact is soon taken over by rotation of the trans-alkene formed in the second step! This reverses the first antarafacial mode, and the net result is that none of these modes survive into the final product cyclo-octatetraene which is now in the valence bond form A rather than B.[4]
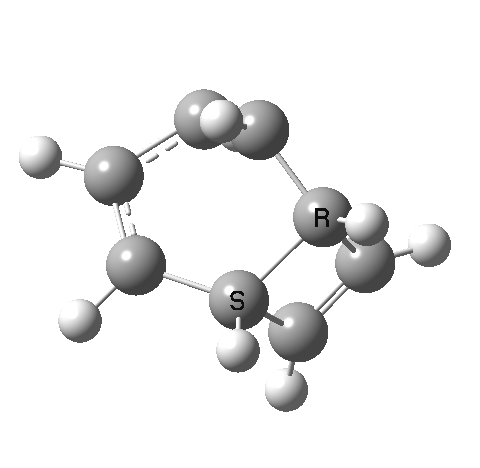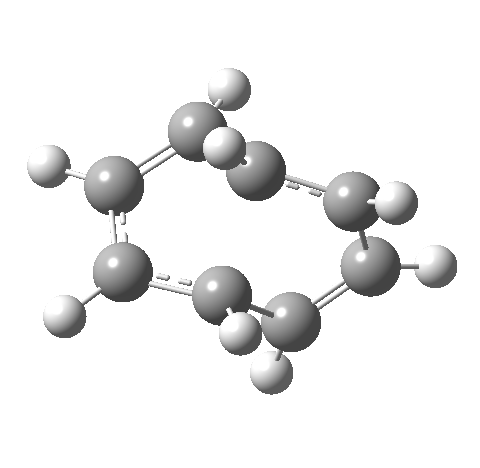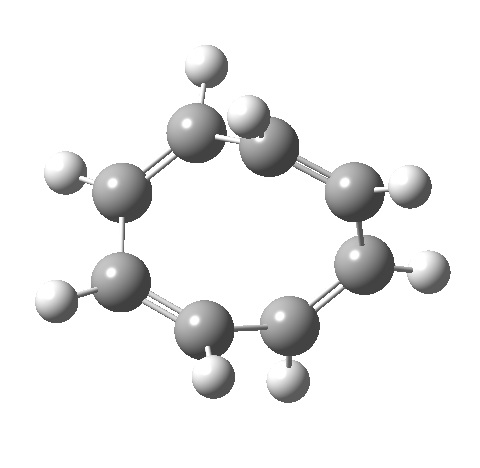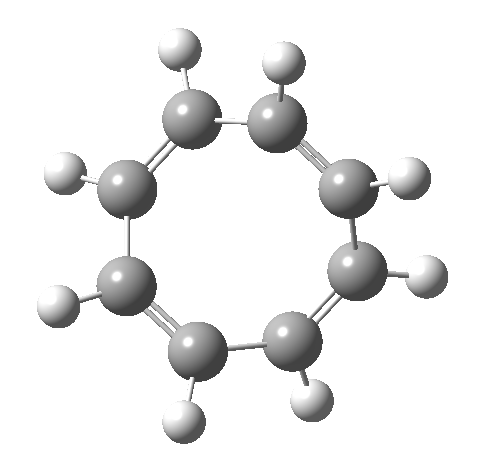


- This second reaction is clearly part of a pericyclic sequence, but quite unlike any I have ever come across previously. In particular, the morphing of the antarafacial mode away from the cyclobutene ring and onto the trans-alkene is indeed quite a novel feature!
- The IRC (above) shows the clear presence of a hidden intermediate (IRC = 2.3), which corresponds to the following (C2-symmetric) species (which might have biradical or zwitterionic character). The central bond threatens to form, but ultimately does not!
So to answer the question posed at the start. Quantum mechanics (but not simply electron counting) suggests the reaction comprises two consecutive electrocyclic ring openings. But the second of these has most unusual features, which perhaps could not have been anticipated. It is not really an electrocyclic, so one could reasonably argue that the answer to the first question posed is in fact none of the above (and I might add that biradical mechanisms have not been considered either).
Perhaps indeed we should start contemplating that the era of simplistic arrow-pushing is coming to an end, and instead we should more routinely start replacing it with quantum mechanical computations. Just a thought!
‡It is perfectly possible that substituents could alter the balance between the cyclo-elimination mode and the two-fold electrocyclic, and resulting in the former being in fact the preferred mode. For example, replacing H by thioformyl (HC=S) flips the mechanism to the 2+2 elimination (it removes that second imaginary frequency).[5]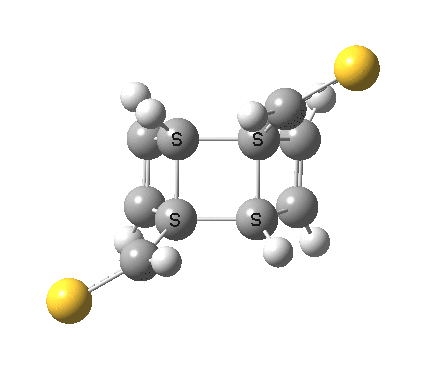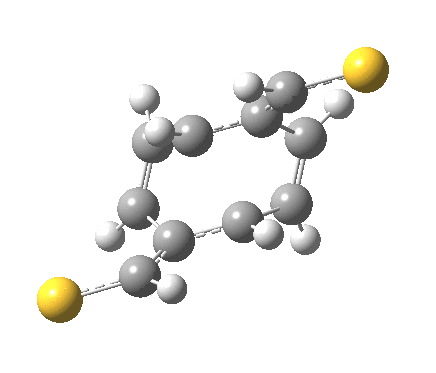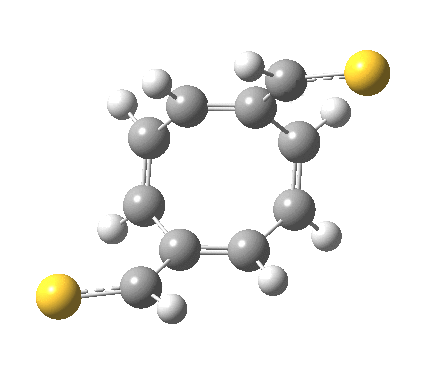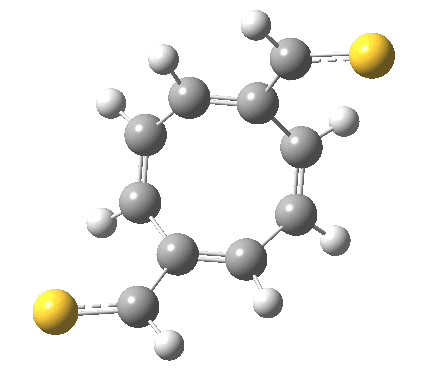
Author
References
- H.S. Rzepa, "Gaussian Job Archive for C8H8", 2014. https://doi.org/10.6084/m9.figshare.899759
- H.S. Rzepa, "Gaussian Job Archive for C8H8", 2014. https://doi.org/10.6084/m9.figshare.899760
- H.S. Rzepa, "Gaussian Job Archive for C8H8", 2014. https://doi.org/10.6084/m9.figshare.899763
- H.S. Rzepa, "Gaussian Job Archive for C8H8", 2014. https://doi.org/10.6084/m9.figshare.899762
- H.S. Rzepa, "Gaussian Job Archive for C10H8S2", 2014. https://doi.org/10.6084/m9.figshare.899809