My previous post related to the aromatic electrophilic substitution of benzene using as electrophile phenyl diazonium chloride. Another prototypical reaction, and again one where benzene is too inactive for the reaction to occur easily, is the catalyst-free bromination of benzene to give bromobenzene and HBr.
![]()

The “text-book” mechanism involves nucleophilic attack by the benzene on the bromine to form a “Wheland intermediate” (the blue arrows) followed in a clear second step by proton removal by the liberated bromide anion (the red arrows). But one group had other ideas[1], proposing in 2011 that the blue and red arrows conflate into a single concerted process which does NOT involve an explicit Wheland intermediate ion-pair. The text-books would have to be re-written! Paul Schleyer (a co-author of the above) recently contacted me about this reaction, noting that no explicit intrinsic reaction coordinate (IRC) had been reported in the 2011 article. Could I run one to establish that the course of this reaction really was concerted and “Whelandless“?
The level of theory used before[1] is rb3lyp/6-311++G(2d,2p)/SCRF=CCl4 (the r is added here, for reasons that will soon become apparent) and the animation[2] is shown below, which is followed by repeating the calculation with addition of a D3-type dispersion correction to the core rb3lyp DFT method.[3] Without dispersion, the final HBr becomes H-bonded to the other Br, but with dispersion it instead forms a π-facial hydrogen bond to the aromatic ring. Even for such a small molecule, one can easily observe the effects of dispersion forces!
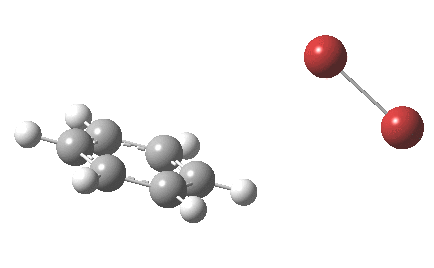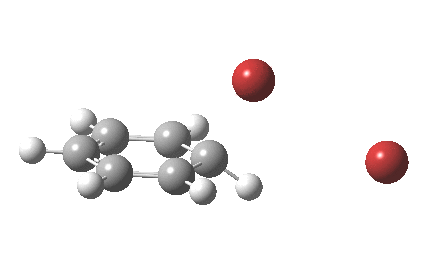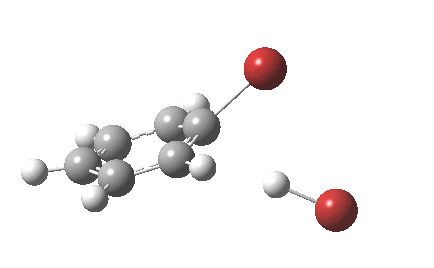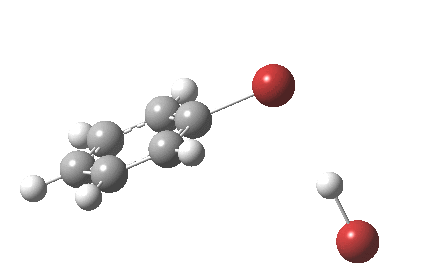
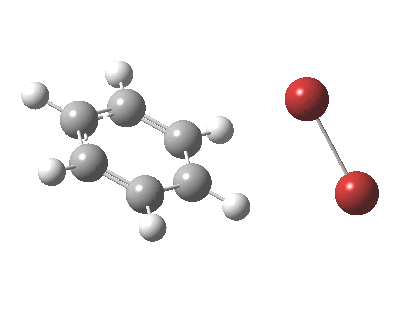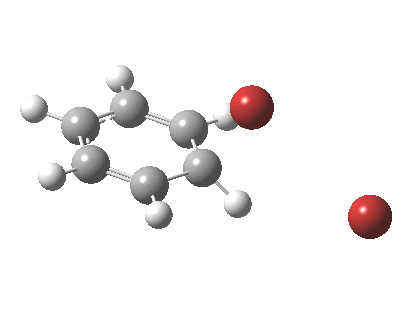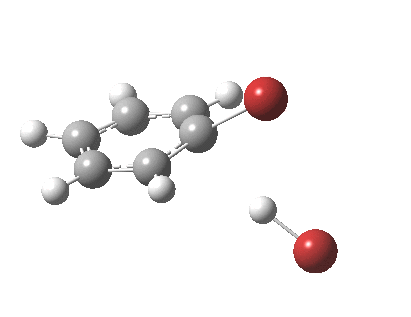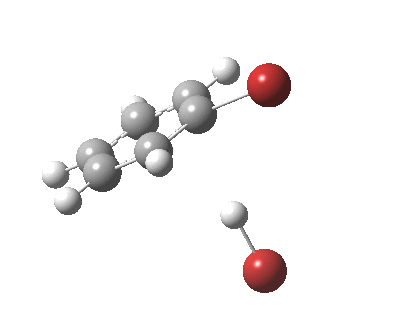


The reaction is indeed concerted, but it is also asynchronous as revealed by the characteristic feature at IRC ~3. We might conclude that the Wheland does make an appearance in this mechanism, but only as a “hidden intermediate“. It is a relay-race with the blue arrows above running first, and then without pause smoothly passing the baton of the reaction to the red arrows. The activation energy is high, commensurate with a reaction that in fact does not take place at normal temperatures.
Boris Galabov (another co-author[1]) then pointed out to me that the spin-restricted wavefunction (r above) at the transition state is unstable with respect to spin unrestriction.[4] This means that some open-shell biradical character is present at least at the transition state if not the entire pathway. So what would happen if the IRC were repeated using ub3lyp instead of rb3lyp? Would allowing for biradical character still retain the concerted nature?
Before showing the results, I have to point out that the uIRC must be done in two stages,‡ the first being the path to the transition state and the second the path down from it to products (the program I use to show the profiles is not capable has errors when splicing the two together). First the upward path[5] (without dispersion) ending at the TS, followed by the path down.[6]
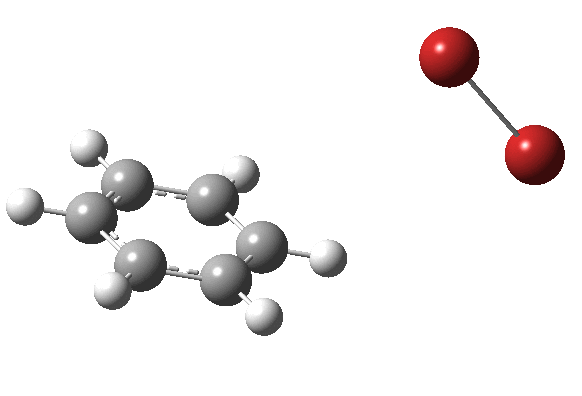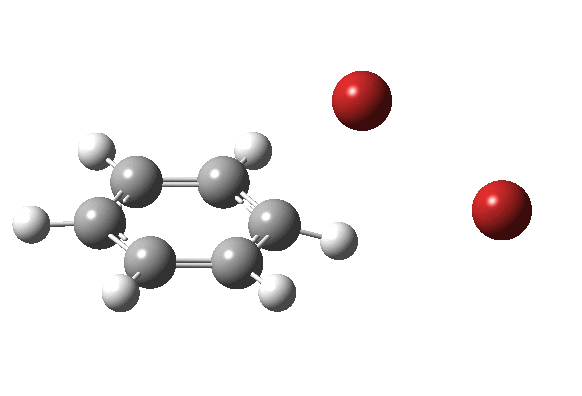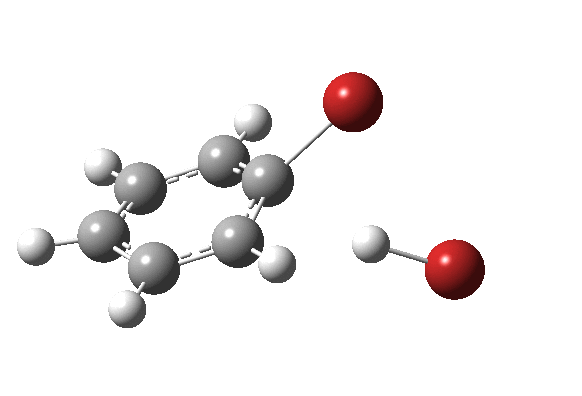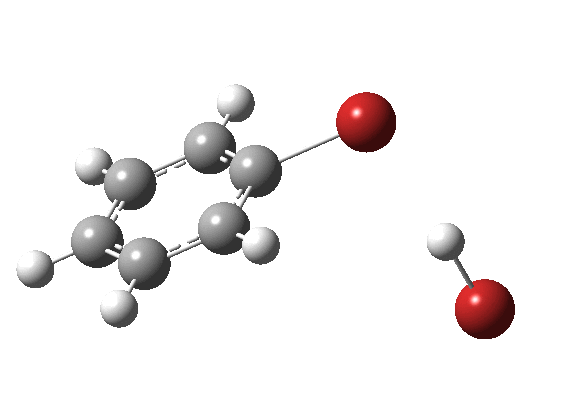
 |
 |
On the approach path, the spin expectation operator <S2> starts at zero but at IRC ~2.0 it becomes non-zero (biradical character forms) and this persists to the transition state and to IRC ~-2 beyond on the downward path before reverting again to a closed shell singlet. In this central region we have what amounts to a “hidden biradicaloid intermediate”. Since the C-Br bond formation and the subsequent C-H bond cleavage are NOT synchronous, we also retain the hidden Wheland characteristics. So this system is perhaps best described as having a “hidden biradicaloid Wheland intermediate“; a double whammy in the vernacular. The non zero value of <S2> lowers the activation barrier from ~42 kcal/mol to ~37 kcal/mol, but it still remains a barrier which is insurmountable at room temperatures.
The bottom line remains: according to this quantum model, the reaction is concerted, as originally claimed.[1]
‡ The technical explanation is as follows. The IRC is started at the TS, and the SCF is converged using a broken-symmetry keyword guess(mix). As the IRC proceeds on the path down to reactant, each step uses the density matrix from the previous step as the initial SCF guess. This ensures that the unrestricted wavefunction remains symmetry broken if that is the lowest energy solution. Before the reactant is reached however, <S2> has collapsed to zero. Then the forward path is started, again from the TS. However, the program continues to use the last density matrix and hence <S2> continues to be zero for this entire path. Hence the reason for performing two separate IRC calculations, to ensure that the correct value of <S2> is achieved on both pathways.
Author
References
- J. Kong, B. Galabov, G. Koleva, J. Zou, H.F. Schaefer, and P.V.R. Schleyer, "The Inherent Competition between Addition and Substitution Reactions of Br<sub>2</sub> with Benzene and Arenes", Angewandte Chemie International Edition, vol. 50, pp. 6809-6813, 2011. https://doi.org/10.1002/anie.201101852
- H.S. Rzepa, "Gaussian Job Archive for C6H6Br2", 2014. https://doi.org/10.6084/m9.figshare.956223
- H.S. Rzepa, "Gaussian Job Archive for C6H6Br2", 2014. https://doi.org/10.6084/m9.figshare.956247
- M.J. Dewar, S. Olivella, and H.S. Rzepa, "MNDO study of ozone and its decomposition into (O2 + 0)", Chemical Physics Letters, vol. 47, pp. 80-84, 1977. https://doi.org/10.1016/0009-2614(77)85311-6
- H.S. Rzepa, "Gaussian Job Archive for C6H6Br2", 2014. https://doi.org/10.6084/m9.figshare.958784
- H.S. Rzepa, "Gaussian Job Archive for C6H6Br2", 2014. https://doi.org/10.6084/m9.figshare.958785
Tags: activation energy, animation, aromatic, Boris Galabov, co-author, electrophilic, lowest energy solution, o/p director of aromatic electrophilic substitution, Paul Schleyer, pence, substitution
Here is a plot I extracted from the spin-unrestricted IRC pathway in the direction transition state to product. It shows the value of <S2> as the reaction proceeds.
The bromination of benzene by Br2 was asynchronous; the C-Br bond was largely formed before the C-H bond started to be abstracted. But just to show that this asynchronicity need not be true, here is the IRC of the reaction of benzene with nitrosyltrifluoroacete (the transition state for which first appeared on this blog).
The characteristic features of a “hidden intermediate” in the gradient norms are absent for this particular reaction; it is a truly concerted synchronous aromatic electrophilic substitution!
Hi Henry,
Very interesting!
And what’s about Br2 dimer as reagent?
May PhHBr+…Br3- be the true intermediate?
In my previous comment, I showed that the electrophilic nitrosation of benzene was both concerted and synchronous. Here, I ask how much it might take to knock it of this perch. Thus a p-amino group (the C-nitrosation of aniline).
Below are shown the IRC animation and the energy and gradient norm profiles. You can see that not only are they different from benzene itself, they are very different by some margin.
The barrier is reduced from ~25 kcal/mol to ~8 kcal/mol, and the synchronicity is almost entirely lost. For this perturbation, the C-N bond formation is almost entirely complete (IRC ~2 to 0) before there is an sign of C-H loss (IRC -6 to -8). The two processes are separated by about 6 IRC units. In other words, the “Wheland intermediate” is almost (but not quite) a stable minimum (IRC 0 to -6). It is “hidden”, but not by much! Thus for benzene, one would predict a hydrogen isotope effect, but for aniline none.
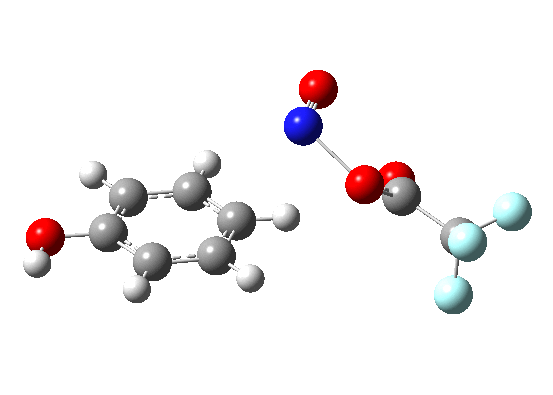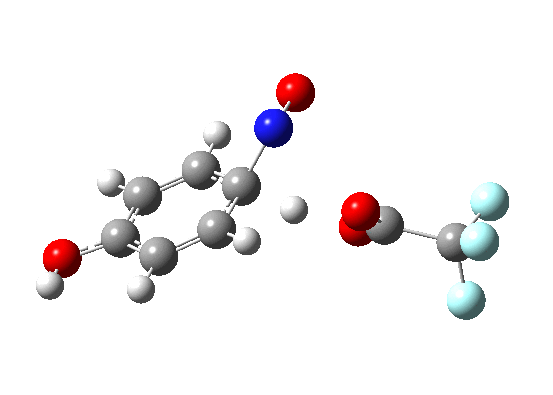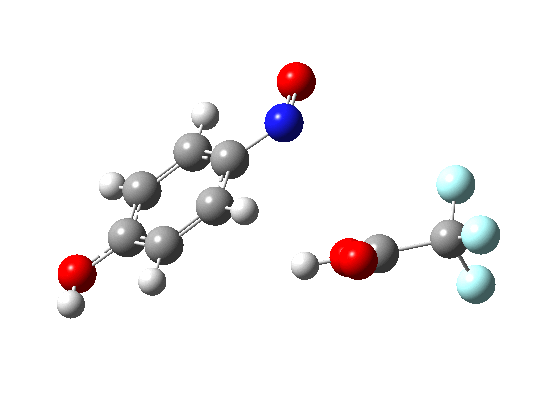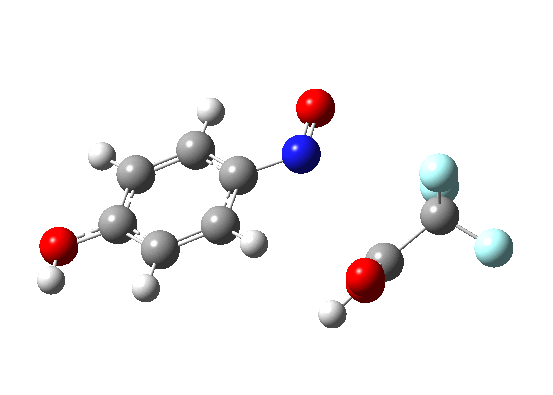
Having covered benzene and p-NH2 benzene (aniline) here I show the one in-between, phenol (p-OH).
Like benzene but unlike aniline, it remains both concerted and synchronous. The barrier is in-between that for benzene and for aniline. I dare say a good free-energy relationship could be conjured up out of these observations.


I thought it might be interesting to show the X-ray structure of the complex formed between bromine and benzene (DOI: 10.1039/b102148f).
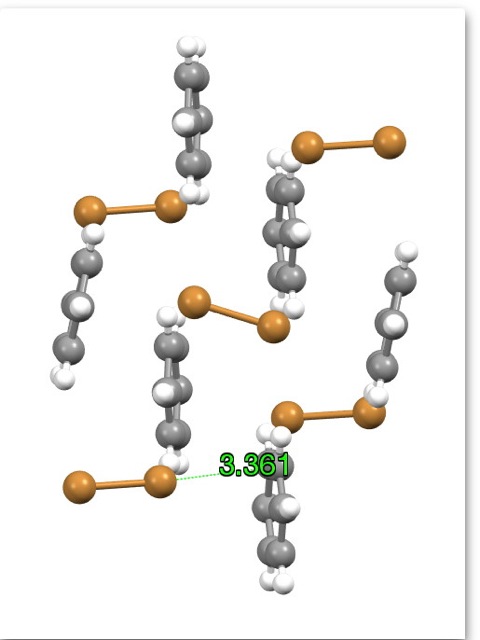
The Br approaches the benzene to a distance of ~ 3.4Å a little shorter than the combined van der Waals distances of ~3.55Å. It is probably a dispersion complex.
What will happen if e do halogenation of benzene with BrCl
[…] Per una discussione differente sulla alogenazione del benzene si legga questo post del chimico Henry Rzepa unitamente a […]