My previous dissection of the mechanism for ester hydrolysis dealt with the acyl-oxygen cleavage route (red bond). There is a much rarer[1] alternative: alkyl-oxygen cleavage (green bond) which I now place under the microscope.

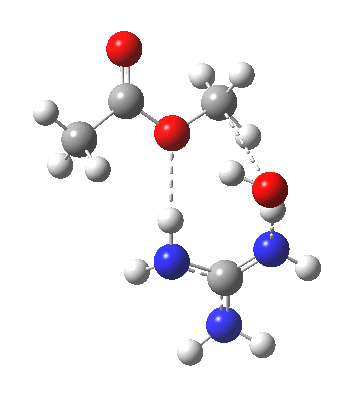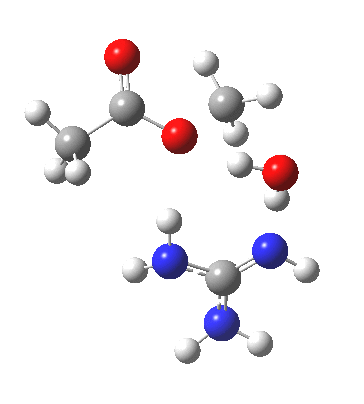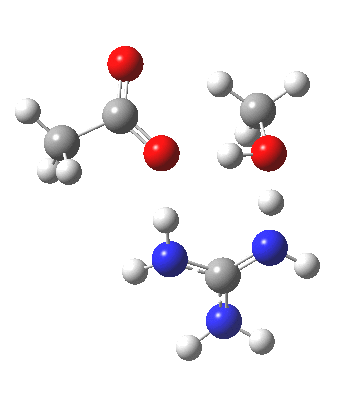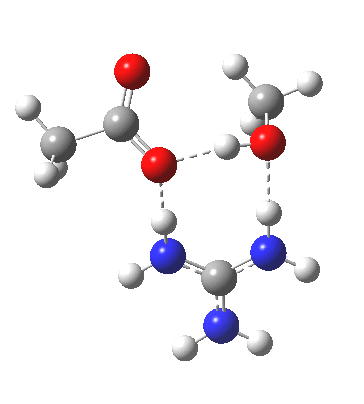
Here, guanidine is used as a general acid/base, which results in a reasonable activation barrier for the hydrolysis (using pure water as the catalyst led to high barriers). What I will call the classical stepwise route is shown above, with charge-separated structures in abundance (particularly at the allyl group, where the possibility of forming a carbocation at this centre is central to the mechanism). My philosophy here is to allow quantum mechanics to decide whether to separate charge or not (in effect, only it is allowed decisions about where electrons are). So one can start with a concerted mechanism in which no formal charges are separated, and by subjecting them to wB97XD/6-311G(d,p)/SCRF=water calculation, decide where and if charges develop.
There are two distinct possibilities; hydrolysis with either retention or inversion of configuration at the alkyl group. The results for the transition states are shown below, with the analogous energy for acyl-oxygen cleavage shown for comparison.
| Relative energies for hydrolysis of Alkyl acetate | |||
|---|---|---|---|
| R | Acyl-oxygen | Alkyl O,inversion | Alkyl-O,retention |
| all H | 0.0 | 15.3 | 42.5 |
| Me | 0.0 | 16.6 | 35.0 |
| Me,Me | 0.0 | 16.3 | 18.2 |
| Me,Me,Me | 0.0 | 16.4 (14.4) | ? |
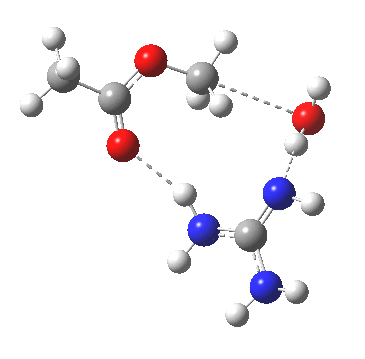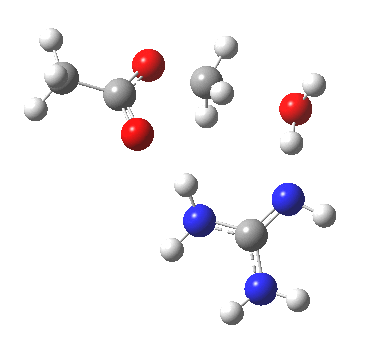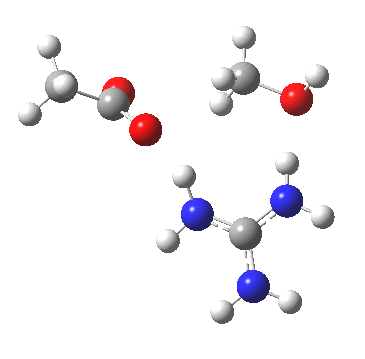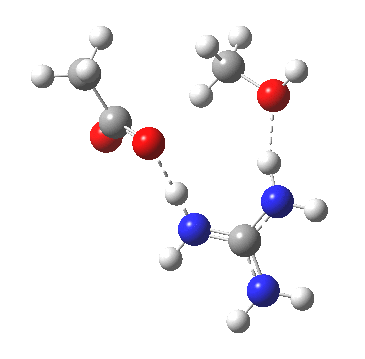
For R1=R2=R3=H and R1=Me,R2=R3=H proceeding with retention of configuration. The IRCs are as below, which reveal a “hidden intermediate” feature (visible as a dip in the gradient norm), which corresponds to a charge-separated zwitterionic intermediate immediately preceding the proton transfer. In other words, the non-charge-separated cyclic/concerted mechanism shown above is “interrupted” by charge separation in a hidden way during, and in an explicit way at the final stage, preferring finally to form the ionic ion-pair rather than neutral acetic acid and guanidine.
 [2] [2] |
 |
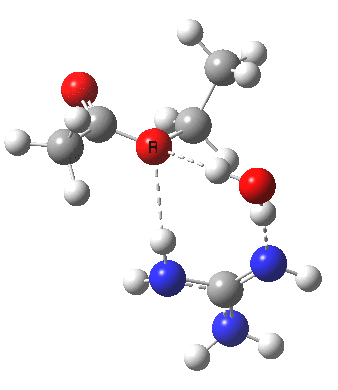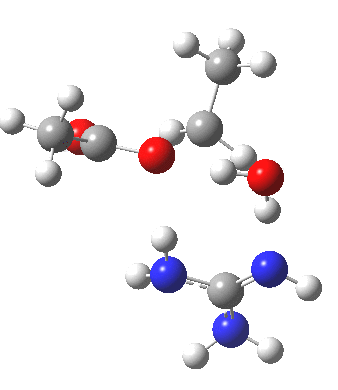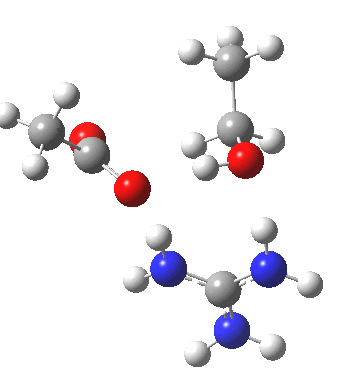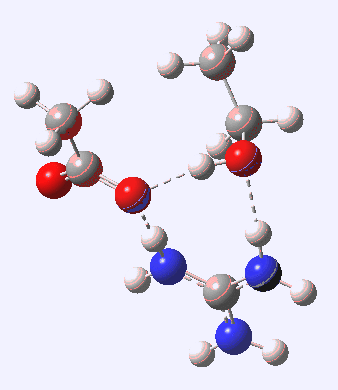 [3] [3] |
 |
 |
|
For R1=R2=Me, R3=H, we have a change. The C-O bond lengths at the solvolysing methyl increase as the substitution at this carbon increases, e.g. 2.2Å (R=H) → 2.4Å (R1=Me) as the transition state becomes more carbocation like. With increasing carbocationic character, the acidity of the adjacent C-H group increases, until with R1=R2=Me, R3=H it has become acidic enough to be abstracted by any close-by base (in this instance, guanidine). Experimentally, the aqueous hydrolysis of t-butyl acetate is known to proceed with alkyl-oxygen cleavage[1]. In the computational model, the solvolysis mechanism has been intercepted by an elimination mechanism: the two potential surfaces under these circumstances are very close and they merge to ensure a different outcome of the reaction. You can see this effect below;
Click for 3D.
The reaction barrier also drops as the degree of substitution at the migrating carbon increases. At time of writing, no TS had been located for R1=R2=R3 (? in table above) but as you can see the trend could easily take it below the energy for acyl oxygen hydrolysis.
A much lower energy route however is apparently available for the alkyl-oxygen solvolysis route. For R1=R2=R3=H, it proceeds much more favourably with inversion of configuration, an intramolecular Sn2 solvolysis in fact.
 |
 |
 |
|
That for R1=R2=R3 shows a qualitative difference, in resembling the mechanism for Sn1 solvolysis of t-butyl chloride in water. In this case the bond O-C bond labelled 2.3 is cleaving, whilst the C-O bond labelled 3.1 has not yet started to form; an apparently classical Sn1 solvolysis. But take a look at the two atoms labelled [1] and [2]; this C-H bond is also set up to be abstracted by an adjacent base (the carboxylate), and indeed an IRC shows the formation of butene (not solvolysis) to be the final outcome.
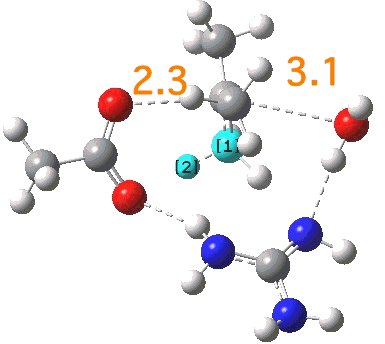
Click for 3D.
Unlike the mechanism involving retention of configuration, the barrier for the inversion route does not change much as the substitution at the carbon increases, remaining above the acyl-oxygen solvolysis for even the t-butyl ester (R1=R2=R3=Me).
To summarise what we might have learnt. Firstly, the mechanism of the apparently simple hydrolysis of alkyl esters of ethanoic (acetic) acid suddenly got much more complicated. It might seem that solvolysis of the O-alkyl bond can proceed with either inversion or retention of configuration at the alkyl carbon; if the latter then the barrier seems to decrease as the stabilisation of the carbocation at this carbon increases. But for both retention and inversion, the mechanistic pathway can easily be subverted by a different reaction involving the formation of an alkene.
One starts to suspect that the model I am using here to study this reaction may be either the wrong kind, or certainly incomplete. In the absence of any explicit water (merely a continuum model acting on its behalf), it seems more basic molecules bound in by hydrogen bonds (guanidine or carboxylate) can take over by acting as bases and abstracting hydrogens from a H-C bond adjacent to the carbocationic centre. In order to redirect the mechanism onto the solvolysis pathway, one probably needs to have a few more explicit water molecules hanging around (so to speak) so as to quickly intercept the forming carbocation, before it can release its proton to the base. In other words, one needs to set up a more statistical model, in which the probability of the desired outcome is in part determined by the probability of having a favourable molecule adjacent to the reacting centre. Who would have thought such a basic prototype for organic chemistry could be so tricky to pin down in a computational model!
Author
References
- C.A. Bunton, and J.L. Wood, "Tracer studies on ester hydrolysis. Part II. The acid hydrolysis of tert.-butyl acetate", Journal of the Chemical Society (Resumed), pp. 1522, 1955. https://doi.org/10.1039/jr9550001522
- H.S. Rzepa, "Gaussian Job Archive for C4H13N3O3", 2013. https://doi.org/10.6084/m9.figshare.663603
- H.S. Rzepa, "Gaussian Job Archive for C5H15N3O3", 2013. https://doi.org/10.6084/m9.figshare.663619
Tags: acetic acid, analogous energy, energy, lower energy route, Reaction Mechanism, Tutorial material
Alkyl oxygen cleavage is not rare at all, since carboxylic esters like p-nitrobenzoates, trifluoroacetates, and even acetates themselves were widely used as leaving groups in the solvolysis days, e.g., as carbocation precursors.
Paul,
Bunton and Wood used isotope labels to prove the O-alkyl bond rather than the O-acyl bond was cleaved in the rate determining step. Was this proof ever applied to any of the carbocation precursors noted above? Or, what other evidence was there that the O-alkyl bond specifically was involved in the rds?
Recently, we used LiI/MeCN to cleave the methyl ester, this reaction should pass through the methyl oxygen cleavage, I- is a much better nucleophile than H2O.