The example a few posts back of how methane might invert its configuration by transposing two hydrogen atoms illustrated the reaction mechanism by locating a transition state and following it down in energy using an intrinsic reaction coordinate (IRC). Here I explore an alternative method based instead on computing a molecular dynamics trajectory (MD).
Posts Tagged ‘simulation’
Reaction coordinates vs Dynamic trajectories as illustrated by an example reaction mechanism.
Monday, March 20th, 2017Chiroptical spectroscopy of the natural product Steganone.
Tuesday, February 10th, 2015Steganone is an unusual natural product, known for about 40 years now. The assignment of its absolute configurations makes for an interesting, on occasion rather confusing, and perhaps not entirely atypical story. I will start with the modern accepted stereochemical structure of this molecule, which comes in the form of two separately isolable atropisomers.

The first reported synthesis of this system in 1977 was racemic, and no stereochemistry is shown in the article (structure 2).[1] Three years later an “Asymmetric total synthesis of (-)steganone and revision of its absolute configuration” shows how the then accepted configuration (structure 1 in this article) needs to be revised to the enantiomer shown as structure 12 in the article[2] and matching the above representation. The system has continued to attract interest ever since[3],[4],[5],[6], not least because of the presence of axial chirality in the form of atropisomerism. Thus early on it was shown that the alternative atropisomer, the (aS,R,R) configuration initially emerges out of several syntheses, and has to be converted to the (aR,R,R) configuration by heating[3]. One could easily be fooled by such isomerism!
References
- D. Becker, L.R. Hughes, and R.A. Raphael, "Total synthesis of the antileukaemic lignan (±)-steganacin", J. Chem. Soc., Perkin Trans. 1, pp. 1674-1681, 1977. https://doi.org/10.1039/p19770001674
- J. Robin, O. Gringore, and E. Brown, "Asymmetric total synthesis of the antileukaemic lignan precursor (-)steganone and revision of its absolute configuration", Tetrahedron Letters, vol. 21, pp. 2709-2712, 1980. https://doi.org/10.1016/s0040-4039(00)78586-8
- E.R. Larson, and R.A. Raphael, "Synthesis of (–)-steganone", J. Chem. Soc., Perkin Trans. 1, pp. 521-525, 1982. https://doi.org/10.1039/p19820000521
- A. Bradley, W.B. Motherwell, and F. Ujjainwalla, "A concise approach towards the synthesis of steganone analogues", Chemical Communications, pp. 917-918, 1999. https://doi.org/10.1039/a900743a
- M. Uemura, A. Daimon, and Y. Hayashi, "An asymmetric synthesis of an axially chiral biaryl via an (arene)chromium complex: formal synthesis of (–)-steganone", J. Chem. Soc., Chem. Commun., vol. 0, pp. 1943-1944, 1995. https://doi.org/10.1039/c39950001943
- B. Yalcouye, S. Choppin, A. Panossian, F.R. Leroux, and F. Colobert, "A Concise Atroposelective Formal Synthesis of (–)‐Steganone", European Journal of Organic Chemistry, vol. 2014, pp. 6285-6294, 2014. https://doi.org/10.1002/ejoc.201402761
A convincing example of the need for data repositories. FAIR Data.
Thursday, January 15th, 2015Derek Lowe in his In the Pipeline blog is famed for spotting unusual claims in the literature and subjecting them to analysis. This one is entitled Odd Structures, Subjected to Powerful Computations. He looks at this image below, and finds the structures represented there might be a mistake, based on his considerable experience of these kinds of molecules. I expect he had a gut feeling within seconds of seeing the diagram.
Computationally directed synthesis: 2,3-dimethyl-2-butene + NO(+).
Saturday, September 6th, 2014In the previous posts, I explored reactions which can be flipped between two potential (stereochemical) outcomes. This triggered a memory from Alex, who pointed out this article from 1999[1] in which the nitrosonium cation as an electrophile can have two outcomes A or B when interacting with the electron-rich 2,3-dimethyl-2-butene.  NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1]
NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1] 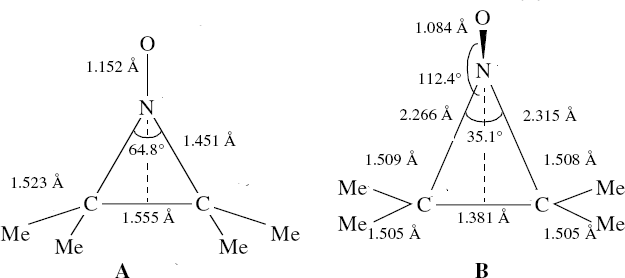 Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest.
Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest. 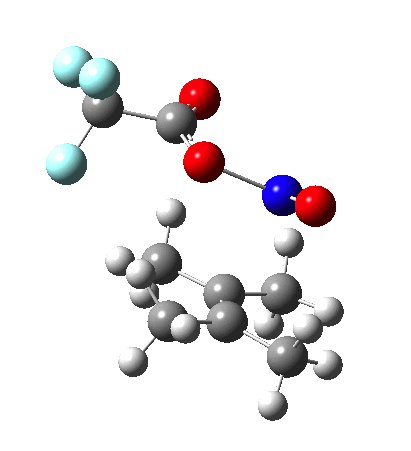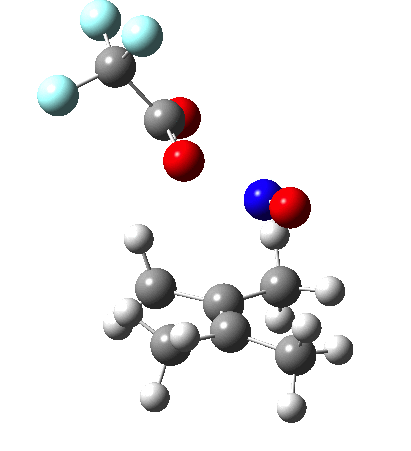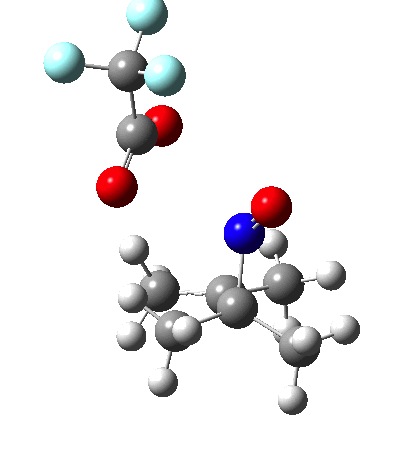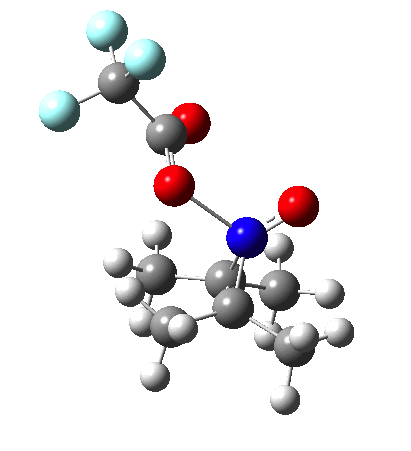

 Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.
Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.  I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate).
I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate). 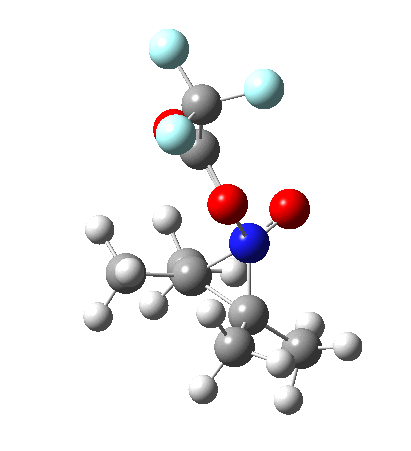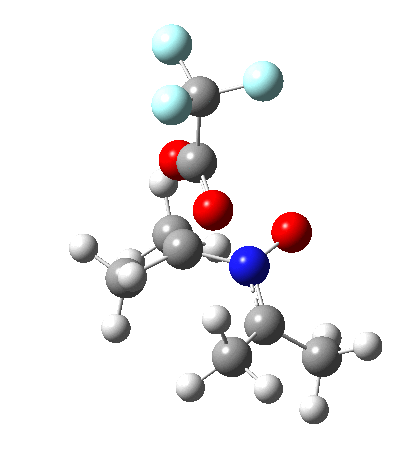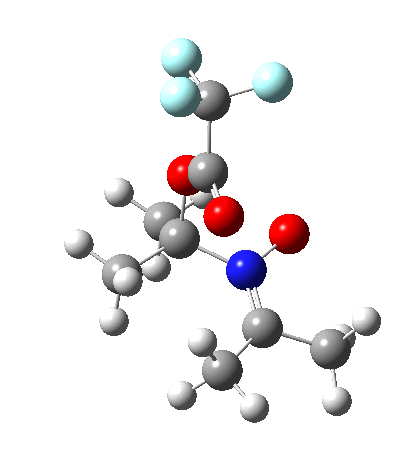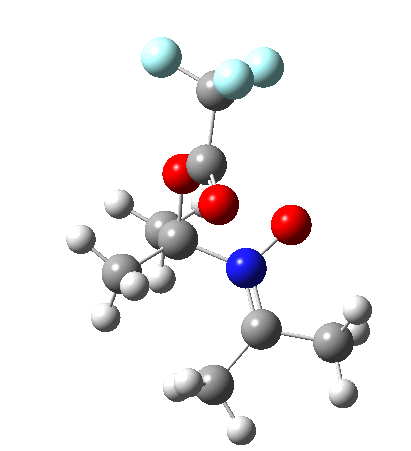
 So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
References
- G.I. Borodkin, I.R. Elanov, A.M. Genaev, M.M. Shakirov, and V.G. Shubin, "Interaction in olefin–NO+ complexes: structure and dynamics of the NO+–2,3-dimethyl-2-butene complex", Mendeleev Communications, vol. 9, pp. 83-84, 1999. https://doi.org/10.1070/mc1999v009n02abeh000995
- H.S. Rzepa, "C8H12F3NO3", 2014. https://doi.org/10.14469/ch/24979
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162797
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162676
Thalidomide. The role of water in the mechanism of its aqueous racemisation.
Saturday, November 10th, 2012Thalidomide is a chiral molecule, which was sold in the 1960s as a sedative in its (S,R)-racemic form. The tragedy was that the (S)-isomer was tetragenic, and only the (R) enantiomer acts as a sedative. What was not appreciated at the time is that interconversion of the (S)- and (R) forms takes place quite quickly in aqueous media. Nowadays, quantum modelling can provide good in-silico estimates of the (free) energy barriers for such processes, which in this case is a simple keto-enol tautomerism. In a recently published article[1], just such a simulation is reported. By involving two explicit water molecules in the transition state, an (~enthalpic) barrier of 27.7 kcal/mol was obtained. The simulation was conducted just with two water molecules acting as solvent, and without any additional continuum solvation applied. So I thought I would re-evaluate this result by computing it at the ωB97XD/6-311G(d,p)/SCRF=water level (a triple-ζ basis set rather than the double-ζ used before[1]), and employing a dispersion-corrected DFT method rather than B3LYP. 
References
- C. Tian, P. Xiu, Y. Meng, W. Zhao, Z. Wang, and R. Zhou, "Enantiomerization Mechanism of Thalidomide and the Role of Water and Hydroxide Ions", Chemistry – A European Journal, vol. 18, pp. 14305-14313, 2012. https://doi.org/10.1002/chem.201202651
Dynamic effects in nucleophilic substitution at trigonal carbon (with Na+).
Thursday, July 19th, 2012In the preceding post, I described a fascinating experiment and calculation by Bogle and Singleton, in which the trajectory distribution of molecules emerging from a single transition state was used to rationalise the formation of two isomeric products 2 and 3. In the present post, I explore possible consequences of including a sodium cation (X=Na+ below) in the computational model.
Computers 1967-2011: a personal perspective. Part 4. Moore’s Law and Molecules.
Friday, October 28th, 2011Moore’s law describes a long-term trend in the evolution of computing hardware, and it is often interpreted in terms of processing speed. Here I chart this rise in terms of the size of computable molecules. By computable I mean specifically how long it takes to predict the geometry of a given molecule using a quantum mechanical procedure.