George Olah passed away on March 8th. He was part of the generation of scientists in the post-war 1950s who had access to chemical instrumentation that truly revolutionised chemistry. In particular he showed how the then newly available NMR spectroscopy illuminated structures of cations in solvents such “Magic acid“. The obituaries will probably mention his famous “feud” with H. C. Brown over the structure of the norbornyl cation (X=CH2+), implicated in the mechanism of many a solvolysis reaction that characterised the golden period of physical organic chemistry just before and after WWII.
Posts Tagged ‘Physical organic chemistry’
George Olah and the norbornyl cation.
Friday, March 10th, 2017Stable “unstable” molecules: a crystallographic survey of cyclobutadienes and cyclo-octatetraenes.
Sunday, March 5th, 2017Cyclobutadiene is one of those small iconic molecules, the transience and instability of which was explained theoretically long before it was actually detected in 1965.[cite]10.1021/ja01092a049[/cite] Given that instability, I was intrigued as to how many crystal structures might have been reported for this ring system, along with the rather more stable congener cyclo-octatetraene. Here is what I found.
A periodic table for anomeric centres, this time with quantified interactions.
Monday, August 8th, 2016The previous post contained an exploration of the anomeric effect as it occurs at an atom centre X for which the effect is manifest in crystal structures. Here I quantify the effect, by selecting the test molecule MeO-X-OMe, where X is of two types:
A periodic table for anomeric centres.
Saturday, August 6th, 2016In the last few posts, I have explored the anomeric effect as it occurs at an atom centre X. Here I try to summarise the atoms for which the effect is manifest in crystal structures.
Anomeric effects at boron, silicon and phosphorus.
Friday, July 1st, 2016
The anomeric effect occurs at 4-coordinate (sp3) carbon centres carrying two oxygen substituents and involves an alignment of a lone electron pair on one oxygen with the adjacent C-O σ*-bond of the other oxygen. Here I explore whether other centres can exhibit the phenomenon. I start with 4-coordinate boron, using the crystal structure search definition below (along with R < 0.1, no disorder, no errors).[cite]10.14469/hpc/696[/cite]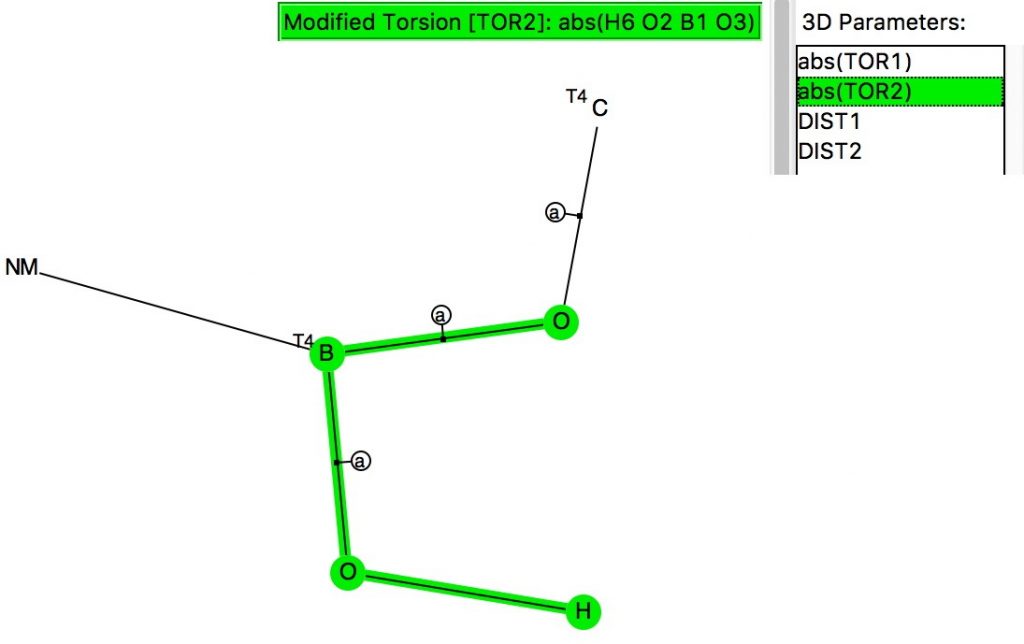
How does an OH or NH group approach an aromatic ring to hydrogen bond with its π-face?
Wednesday, June 22nd, 2016I previously used data mining of crystal structures to explore the directing influence of substituents on aromatic and heteroaromatic rings. Here I explore, quite literally, a different angle to the hydrogen bonding interactions between a benzene ring and OH or NH groups.
The geometries of 5-coordinate compounds of group 14 elements.
Monday, May 30th, 2016This is a follow-up to one aspect of the previous two posts dealing with nucleophilic substitution reactions at silicon. Here I look at the geometries of 5-coordinate compounds containing as a central atom 4A = Si, Ge, Sn, Pb and of the specific formula C34AO2 with a trigonal bipyramidal geometry. This search arose because of a casual comment I made in the earlier post regarding possible cooperative effects between the two axial ligands (the ones with an angle of ~180 degrees subtended at silicon). Perhaps the geometries might expand upon this comment?
A molecular balance for dispersion energy?
Sunday, February 7th, 2016The geometry of cyclo-octatetraenes differs fundamentally from the lower homologue benzene in exhibiting slow (nuclear) valence bond isomerism rather than rapid (electronic) bond-equalising resonance. In 1992 Anderson and Kirsch[cite]10.1039/P29920001951[/cite] exploited this property to describe a simple molecular balance for estimating how two alkyl substituents on the ring might interact via the (currently very topical) mechanism of dispersion (induced-dipole-induced-dipole) attractions. These electron correlation effects are exceptionally difficult to model using formal quantum mechanics and are nowadays normally replaced by more empirical functions such as Grimme's D3BJ correction.[cite]10.1002/jcc.21759[/cite] Here I explore aspects of how the small molecule below might be used to investigate the accuracy of such estimates of dispersion energies.