A quartet of articles has recently appeared on the topic of cyclobutadiene.[1],[2],[3],[4]. You will find a great deal discussed there, but I can boil it down to this essence. Do the following coordinates (obtained from a (disordered) previously published[5] x-ray refinement) correspond to a van der Waals complex of 1,3-dimethyl cyclobutadiene and carbon dioxide, or do they instead represent a covalent interaction between these two components resulting in a compound with the chemical name 2-oxabicyclo[2.2.0]hex-5-en-3-one (i.e. not a cyclobutadiene)?
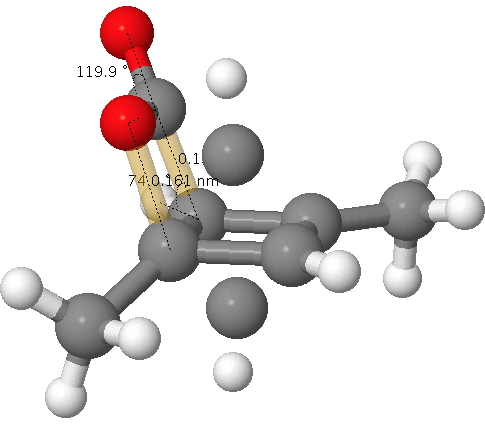
Click for 3D. The unconnected atoms are the result of disordered partial occupancy.
The two bonds to concentrate on are shown in gold; a O…C pair with a distance of 1.61Å as obtained from the x-ray refinement and a C…C pair with a distance of 1.5Å (and if you want to go further, the O=C=O bond angle). I list below values obtained from the wonderful Webelements site. Using these values, this makes a van der Waals O…C contact 3.22Å and a C…C contact 3.40Å and covalent values of respectively 1.38Å and 1.5Å.
| Element | Covalent radius, Å | van der Waals radius |
| C | 0.75 | 1.70 |
| O | 0.63 | 1.52 |
According to chemistry convention, we classify the interaction between a pair of atoms according to which category best fits the observed distance. So this should allow you to decide if the molecule is a van der Waals complex of 1,3-dimethyl cyclobutadiene and carbon dioxide or the covalent system 2-oxabicyclo[2.2.0]hex-5-en-3-one.
Oh, if the observed O…C pair with a distance of 1.61Å does not seem to perfectly fit either category above, one of the quartet of articles above[1] offers the explanation of an unusual π-anomeric effect lengthening the C…O bond in 2-oxabicyclo[2.2.0]hex-5-en-3-one slightly beyond the standard covalent distance. Of course, if the system were to be a van der Waals complex, that explanation cannot apply.
Author
References
- H.S. Rzepa, "A Computational Evaluation of the Evidence for the Synthesis of 1,3‐Dimethylcyclobutadiene in the Solid State and Aqueous Solution", Chemistry – A European Journal, vol. 19, pp. 4932-4937, 2013. https://doi.org/10.1002/chem.201102942
- M. Shatruk, and I.V. Alabugin, "Reinvestigation of “Single‐Crystal X‐ray Structure of 1,3‐dimethylcyclobutadiene”", Chemistry – A European Journal, vol. 19, pp. 4942-4945, 2013. https://doi.org/10.1002/chem.201103017
- Y. Legrand, D. Dumitrescu, A. Gilles, E. Petit, A. van der Lee, and M. Barboiu, "A Constrained Disorder Refinement: “Reinvestigation of “Single‐Crystal X‐ray Structure of 1,3‐Dimethylcyclobutadiene” by M. Shatruk and I. V. Alabugin”", Chemistry – A European Journal, vol. 19, pp. 4946-4950, 2013. https://doi.org/10.1002/chem.201203234
- Y. Legrand, D. Dumitrescu, A. Gilles, E. Petit, A. van der Lee, and M. Barboiu, "Reply to A Computational Evaluation of the Evidence for the Synthesis of 1,3‐Dimethylcyclobutadiene in Solid State and Aqueous Solution—Beyond the Experimental Reality", Chemistry – A European Journal, vol. 19, pp. 4938-4941, 2013. https://doi.org/10.1002/chem.201203235
- Y. Legrand, A. van der Lee, and M. Barboiu, "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix", Science, vol. 329, pp. 299-302, 2010. https://doi.org/10.1126/science.1188002
Tags: crystallography, cyclobutadiene, Waals complex, X-ray
Trying to prove pretty much anything from the disordered part of a crystal structure is an exercise in futility. You could always model it in a number of different ways, even if you made the assumption that it was the same chemical species in each case.
For those that do not know, the reported structure for any crystal structure represents the average over all the symmetry equivalent positions in the crystal. As soon as you have disorder, the problem is that there can be many different ways to get the same average. Most of the time it is the same chemical species in different orientations (such as a “rotating” tert-butyl group), but this does not have to be the case. It could be that this area of the structure contains chemically different species throughout the crystal (e.g. sometimes a tert-butyl and sometimes an iso-propyl).
To summarise, one should always be very wary of drawing significant conclusions involving disordered parts of crystal structures.
Sometimes a “significant” conclusion has to be inferred for the apparently straightforward purpose of indexing a database, as in for example one that supports sub-structure searching. Here, the so-called atom-atom connection table is used to construct the inverted database, and the decision has to be taken at least on what atoms are connected or not. In this article for example, I discuss how searches of the CCDC crystallographic database for compounds with weak S…S contacts depend very much on whether one specifies the structure with, or without an explicit S-S bond. To be safe, one has to try both, and combine them with a Boolean OR operator.
For the system discussed in the post above therefore, one would have to know whether the carbon dioxide had been indexed with or without an explicit bond to the cyclobutadiene part. In fact, if one searches CCDC for 1,3-dimethylcyclobutadiene, one gets exactly zero hits, because in fact the crystal structure first reported in ref. 5 has actually been indexed WITH a covalent bond between the two molecules, thus making it no longer 1,3-dimethylcyclobutadiene but 2-oxabicyclo[2.2.0]hex-5-en-3-one. Defining this latter molecule as the search term does retrieve the reference.
I might add that one can also do searches based on atom-atom distances, but in comparison to those based purely on connection tables, they are very slow. I doubt one could specify more than a few such distances before the search becomes impossibly slow.
[…] But does it have what might be called a conventional chemistry? Other highly reactive species (cyclobutadiene is a well-known example) can often be tamed by trapping as a ligand coordinated to a metal and so […]
I note with interest this article: 10.1021/ja407398w where a claim is made for the shortest non-bonded contact distance for any crystallographically characterised compound. This was for a non-bonded distance of ~1.5Å between two hydrogen atoms. The origin of this contact was claimed as extreme compression by the outer scaffold of the molecule.
Of course, you may wish to consider whether the structure reported by Barboiu et al is another claimant for the shortest non-bonded contact distance, in this case between two carbon atoms rather than two hydrogen atoms, also with a distance of ~1.5Å. If the C-C claim were to be verified, that would be a very much more spectacular result than the same distance for H…H.