I mentioned in the last post that one can try to predict the outcome of electrophilic aromatic substitution by approximating the properties of the transition state from those of either the reactant or the (presumed Wheland) intermediate by invoking Hammond’s postulate[1]. A third option is readily available nowadays; calculate the transition state directly. Here are the results of exploring this third variation.

I am going to use the model shown above, which is actually the relatively unusual electrophile nitrosium trifluoracetate. My reasons for this strange selection are:
- I prefer the complete model, with counter-ion. In this instance, we leave open the option of whether the reagent reacts via an ion-pair or whether it involves a concerted process involving covalency at any stage for the O=N…O bond.
- To make this model more realistic, we are going to add a continuum solvent field (dichloromethane) to allow any (partial) ion-pair character to develop.
- The acetate counter-ion is also retained in order to allow the proton removal to occur, either concurrently with the formation of a C-N bond or (pre- or post) successively with it.
- This combination does allow for a properly characterised transition state to be located and an intrinsic reaction coordinate can then be used to probe for the nature of the pathway.
- This model is then applied to three positions around the pyrrole ring, including the nitrogen itself (position 1). The known outcome of course is that the electrophile substitutes in the 3-position.
The mechanism can either be a more conventional stepwise nucleophilic/electrophilic push-pull (blue + green arrows) or it has the potential of avoiding the formation of any (Wheland) intermediate by instead being a concerted (red + green arrows) process. We will leave the detailed timing of these arrows to the quantum mechanics to settle. The results (ωB97XD/6-311G(d,p)/SCRF=dichloromethane) are as follows (relative energies in kcal/mol).
Substitution at the nitrogen (1-position) is the clear winner in terms of the free energy of activation (ΔG‡, kinetic control) but the clear looser in terms of the free energy of reaction (thermodynamic control).
| Position | Transition state | Product |
| 1 | -4.93 | 10.62 |
| 2 | 1.96 | 4.86 |
| 3 | 0.0 | 0.0 |
Time to take a detailed look at the three transition states located and their intrinsic reaction coordinates.
- The IRC profile for the N-reaction is a nice example of a concerted reaction (the equivalent red + green arrows above) in which the trifluoracetate firstly heterolyses off the nitrosonium cation to form an ion-pair, and then as a basic anion, it abstracts the relatively acidic N-H proton. Only then does the N-NO bond fully form to quench the ion-pair. The overall barrier to this process is only ~9 kcal/mol. This detailed choreography is certainly not a variation I have ever seen described in any text-book!‡
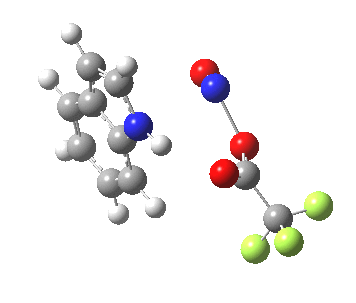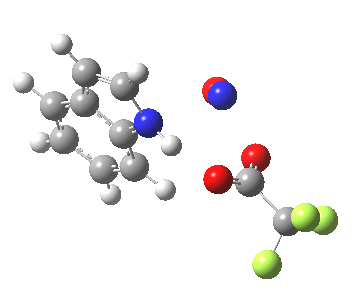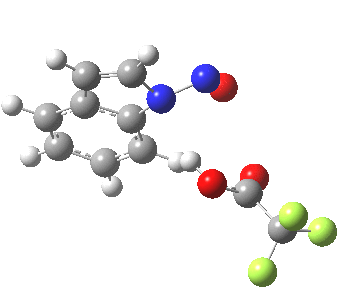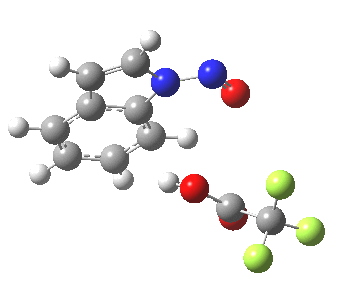

- In contrast, the IRC for substitution at the 3-position reverses the order of C-N formation and C-H removal, the latter now happening at the end (IRC ~ -5) rather than at the start. As before, the process is however still concerted, with no formation of an actual Wheland intermediate at any stage. This makes the Hammond-based prediction of the transition state properties by extrapolating those of such a presumed intermediate rather tenuous if the intermediate in question actually has no existence on the potential energy surface!
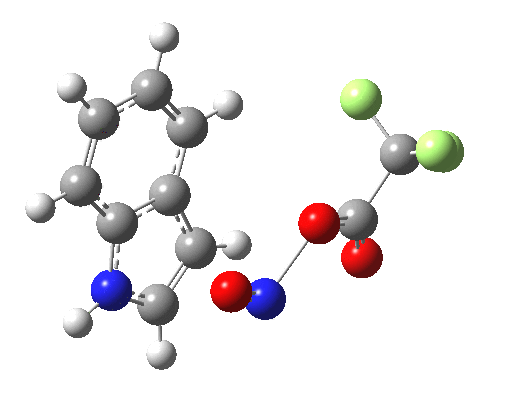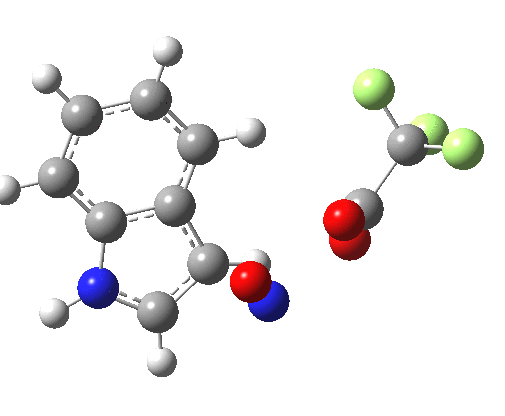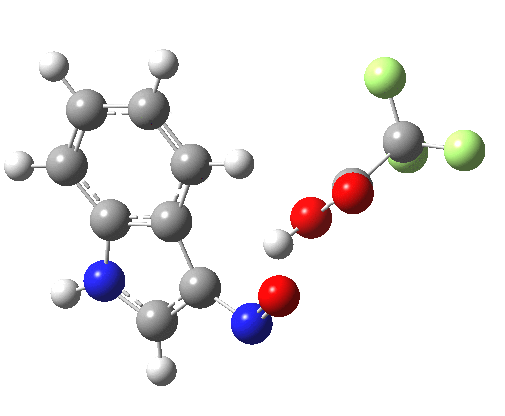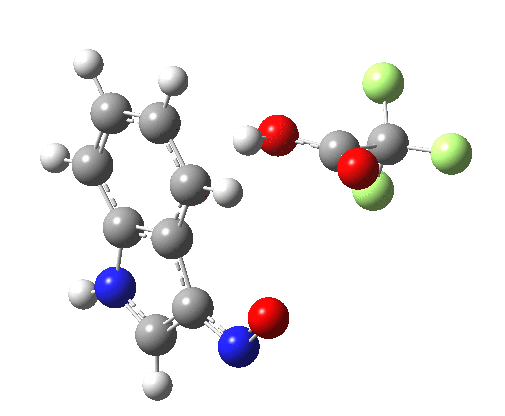

- Yet another surprise in store for reaction at the 2-position. Although the transition state itself has the form expected and the IRC leads down from this TS to the expected 2-substituted product, the trifluoracetate counter-ion adopts a different role by being enticed away from a cyclic geometry to instead form a strong hydrogen bond to the N-H proton. This means that the start point is no longer the covalent nitrosyltrifluoroacetate, but instead an ion-pair involving an actual Wheland intermediate at the 3-position! So the existence or otherwise of this intermediate very much depends on where the counter-ion is. I would again remind that this counter-ion rarely has much of a role (if any) to play in text-book analyses of this reaction. And now the reaction becomes one involving a migration of the NO group from the 3-Wheland intermediate to the 2-position, followed by proton-removal from that position. The barrier (~15 kcal/mol) is however higher than the others, and so this variant pathway is not actually observed.

The actual outcome (3-position) emerges as the clear thermodynamic winner, but 1-substitution as the (reversible?) kinetic preference. This does raise one intriguing question: might electrophilic substitution of indole in the 3-position actually arise from this initial kinetically controlled 1-substitution followed by some form of rearrangement to the most stable thermodynamic 3-product? I have not identified such a route, which may well be mediated by the position of the trifluoracetate component (and the nature of the solvent and its ability to stabilize ion-pairs).
I am however encouraged that this exploration of transition states has if nothing else introduced some new ideas. I do worry that much organic chemistry continues to be taught against the “text-book” interpretations, and we do need to identify conduits for new ideas to ensure that the core of organic chemistry continues to be vibrant.
Postscript: If you inspect the tail end of the IRC for the 3-indole substitution, you will see the formation of trifluoroacetic acid by proton abstraction from the 3-position. This tail involves a gradual drifting of this acid (IRC ~-10 to -18) to take up a new position over the 4-carbon of the indole by the formation of a π-facial bond. This more or less coincides with the shape of the molecular electrostatic potential of the product in that region (below).
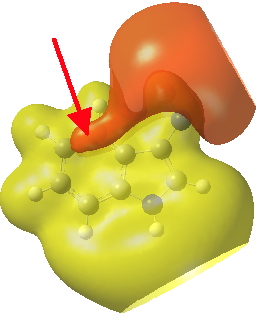
Click for 3D.
‡ A concerted process for aromatic electrophilic substitution of benzene by the nitrosonium cation has been reported[2], but here the proton transfer occurs AFTER the C-N=O bond is formed.
Author
References
- G.S. Hammond, "A Correlation of Reaction Rates", Journal of the American Chemical Society, vol. 77, pp. 334-338, 1955. https://doi.org/10.1021/ja01607a027
- S.R. Gwaltney, S.V. Rosokha, M. Head-Gordon, and J.K. Kochi, "Charge-Transfer Mechanism for Electrophilic Aromatic Nitration and Nitrosation via the Convergence of (ab Initio) Molecular-Orbital and Marcus−Hush Theories with Experiments", Journal of the American Chemical Society, vol. 125, pp. 3273-3283, 2003. https://doi.org/10.1021/ja021152s
[…] the metal lithium is a relative mechanistic newcomer, compared to the pantheon of knowledge on aromatic electrophilic substitution. Investigating the mechanism using quantum calculations poses some interesting challenges, ones I […]
[…] two previous explorations of aromatic substitutions have involved an electrophile (NO+ or Li+). Time now to look at a nucleophile, representing nucleophilic aromatic substitution. The […]