The transient π-complex formed during the “[5,5]” sigmatropic rearrangement of protonated N,O-diphenyl hydroxylamine can be (formally) represented as below, namely the interaction of a six-π-electron aromatic ring (the phenoxide anion 2) with a four-π-electron phenyl dication-anion pair 1. Can one analyse this interaction in terms of aromaticity?

I showed previously that the interaction between these two components involves the stabilising overlap (donation) of a filled orbital on 2 with an empty (acceptor) orbital on the dication-anion pair 1. So what does the interaction of a six-electron (and hence 4n+2 Hückel aromatic) donor ring with a four-electron (and hence formally a Hückel anti-aromatic) acceptor ring lead to? To find out, I carried out a QTAIM analysis of the ring- and bond-critical points in the topology of the computed electron density of complex, and then evaluated the NICS (nucleus-independent-chemical shift) NMR probe at these points. First, the QTAIM analysis. Green=bond critical points, red=ring and blue=cage.
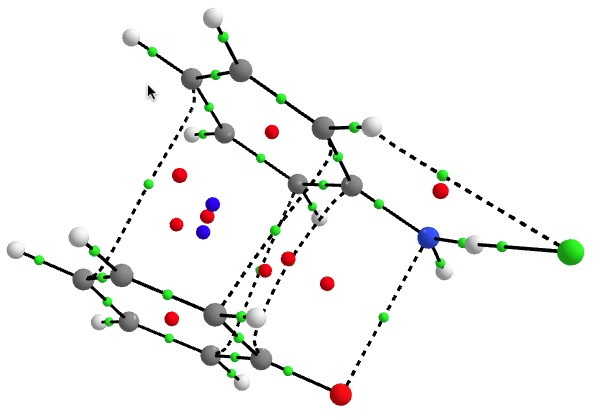
The NMR analysis (ωB97XD/6-311G(d,p)/SCRF=water) is shown below. This is for a closed shell wavefunction, which does not include contributions from any open shell biradical singlet.
Click for 3D.
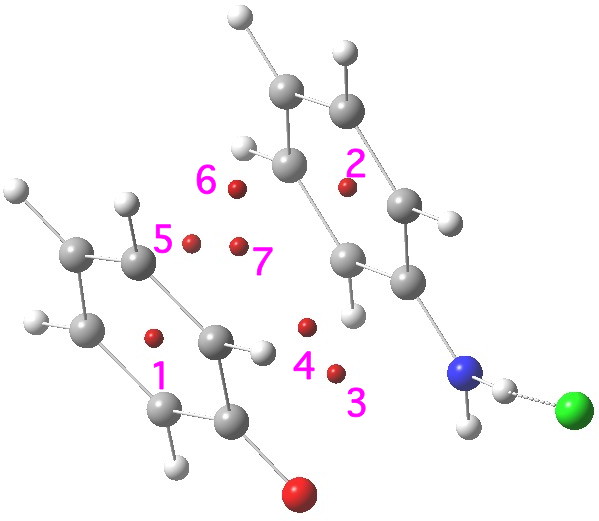
- This is the ring centroid of the phenoxide anion 2, and would normally be expected to show a highly diatropic NICS value indicative of ring aromaticity. The value computed for the complex is -0.9 ppm, which is not aromatic!
- This is the ring centroid of the (nominally) antiaromatic 1, and has a value of -5.9 (mildly aromatic; benzene itself on this scale is about -10 ppm). Neither ring is behaving as might be indicated they should prior to their forming the π-complex.
- The remaining points all lie in plane between the two rings; they are unique to the π-complex itself. Point # 3 has a NICS of -14.0 ppm; it is ~located at the centroid of the C=O and C=N bonds.
- This point has NICS -17.1 ppm, being the most highly diatropic of the seven computed.
- This, -12.1, and
- the next -13.2 are ring points lying between the 3,3′ carbons of either ring.
- This point is the (defining) centroid of the whole complex and has NICS -15.7 ppm.
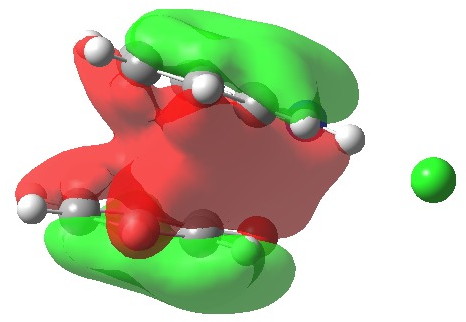
This reveals that neither individual ring of the complex sustains a a diatropic ring current, but that the region between the two rings, one that defines the π-complex itself, is very highly diatropic. The most simple way of looking at it is that the two rings coming together has created an aromatic complex (I remind again that this is in the closed-shell picture of this system, allowing partial biradical character may influence this). To illustrate this holistic aspect, I show below the most stable of the π-MOs (this MO in fact resembles to remarkable degree the lowest π-MO of ferrocene which can be used to illustrate the 18-electron filled shells of the iron at the centre). in fact this is one of seven π-MOs that can be identified, making the system a 14 (certainly 10)-π-electron aromatic (the extra electrons come from the oxygen of 2 and the C=N region of 1).
Click for 3D.
It is most amusing (which is how Michael Dewar might have stated it) that such an unpretentious molecule as PhNHOPh could reveal such surprises. It is also noteworthy that Dewar championed the concept of using aromaticity to determine selection rules for pericyclic reactions, and so he would perhaps have appreciated that the π-complex he suggested for the benzidine pericyclic rearrangement might have its own unique aromatic character.
Author
Tags: chemical, Michael Dewar
[…] on this basis (using this occupancy as the single reference; indeed only state) resulted in the conclusion that the complex was aromatic. The diatropicity (~magnetic aromaticity) was strongest in region between the two stacked rings, […]