Astronomers who discover an asteroid get to name it, mathematicians have theorems named after them. Synthetic chemists get to name molecules (Hector’s base and Meldrum’s acid spring to mind) and reactions between them. What do computational chemists get to name? Transition states! One of the most famous of recent years is the Houk-List.
In the last 12 years or so, the area of enantioselective organocatalysis has blossomed, and an important example involves the asymmetric amino acid (S)-proline (below, shown in green). As its enamine derivative (below, shown in blue), it can catalyse the aldol condensation with an aldehyde or ketone to form two new adjacent stereogenic centres resulting from C-C bond formation (shown below as (R) and (S) as attached to the carbons connected to the red bond).
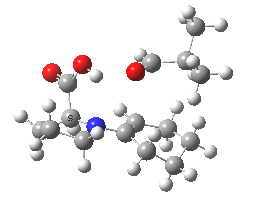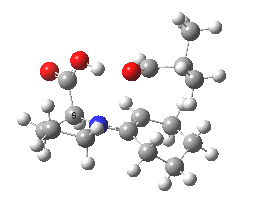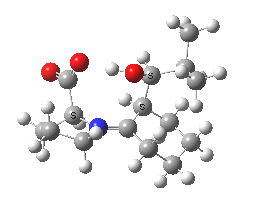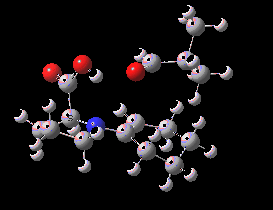
The Houk-List transition state was located for this reaction, and as a useful model for rationalising the stereospecificity of this reaction it has become justly famous (although to be fair, other models have also been proposed). The challenge is to identify the factors selecting for just one stereoisomer (S,R in this case) over the other three (a similar challenge is described in this post for the heterotactic polymerisation of lactide). Houk, List and co-workers constructed their model (the example shown below is for R=isopropyl) as follows.
Houk-List transition state. Original geometry.
Well, this transition state is now nine years old. Unlike asteroids, or mathematical theorems, or indeed molecules and their reactions, a transition state is a slightly more ephemeral object. Its features and properties do rather depend on the particular quantum model used to construct it. There is one feature of the model, necessary in 2003, but no longer so in 2012. This was the use of a gas-phase optimised geometry, augmented at that geometry with a so-called single-point solvation energy correction. Nowadays, the solvation correction is included in the energy used in the geometry optimisation, which now properly reflects the effect of the solvation. Re-optimisation with this inclusion, at the ωB97XD/6-311G(d,p)/SCRF=dmso level thus updates the original Houk-List geometry.
(S,R) Houk-List transition state, updated geometry. Click for 3D
Which brings us to the main point; what is the origin of the diastereoselectivity? An NBO analysis can compare the total steric exchange energy (due to Pauli bond-bond repulsions) of the four isomers, which turns out to be respectively 1214, 1221, 1235 and 1229 kcal/mol. In other words, the favoured isomer has the smallest steric exchange energy. Of course this one term is not the only contributing factor, and a more elaborate analysis will no doubt provide further insight.
So an update to the Houk-List transition state reveals the general characteristics are intact and it is still a very useful model for analysing stereoselectivity in proline organocatalysis.
Postscript: The Intrinsic reaction coordinate (for (S,S) ) is shown below.
In the mid to late 1990s as the Web developed, it was becoming more obvious…
I have written a few times about the so-called "anomeric effect", which relates to stereoelectronic…
The recent release of the DataCite Data Citation corpus, which has the stated aim of…
Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this…
In the late 1980s, as I recollected here the equipment needed for real time molecular…
On 24th January 1984, the Macintosh computer was released, as all the media are informing…
{kind=link}
{kind=link}
{kind=link}
View Comments
Of course 9 years have passed since the original studies and there has been a huge progress in theoretical chemistry and computational power, so there is no need to stick to the old methods. But I'm nevertheless curious if the changes of the TS geometry (and therefore energies) are mainly because of optimization using solvation model or better functional and basis set?
Did you try optimizing the TS using ωB97XD/6-311G(d,p) without SCRF? If yes, how big are the differences?
I have not done all the control experiments, but my experience is that the differences are largely due to the inclusion of solvation as part of the geometry optimisation. These transition states have dipole moments of around 10D, and at this magnitude, (continuum) solvation does start affecting the computed geometries. Of course, adding explicit solvent, by forming hydrogen bonds to the ends of the dipole can also affect the geometry.
It would also be true to say that for the explicit case of proton transfers, these can be VERY sensitive to the functional, and to how correlation is treated. For such a simple reaction, it can be a complicated one to get right, as perhaps this post suggests.