The title comes from the abstract of an article[1] analysing why Biotin (vitamin B7) is such a strong and effective binder to proteins, with a free energy of (non-covalent) binding approaching 21 kcal/mol. The author argues that an accumulation of both CH-π and CH-O together with more classical hydrogen bonds and augmented by a sulfur centered hydrogen bond, oxyanion holes and water solvation, accounts for this large binding energy. 
Here, I thought I would present a visualisation of the surroundings of biotin using the method of NCI (non-covalent-interaction) analysis, which looks at the behaviour of the electron density in the “weak” (i.e. non-covalent) regions of the biotin. This provides a more objective measure of the important interactions, independent of what we might consider important by virtue of having labels attached (such as e.g. “hydrogen bond”).
- I started by getting the coordinates of streptavidin (DOI: 10.2210/pdb3RY2/pdb) a protein where biotin has been co-crystallised.[2]
- Loaded into the CCDC Mercury program, I selected the molecule biotin itself and then added to the selection its close contacts with various groups in the streptavidin protein. These additions were truncated and capped with a methyl group to allow a wavefunction for the assembly to be calculated.
- Hydrogens were then added to this structure to complete atom valencies, using “idealised” positions and ensuring that when rotamers were possible, they were set up to form hydrogen bonds.
- A calculation (DOI: 10.14469/hpc/9982 at the ωB97XD/Def2-TZVPP/SCRF=water level) was performed.
- The heavy atom coordinates (i.e. not hydrogens) are unaltered from the X-ray structure. Since atom positions as measured by X-ray diffraction and as computed using a DFT procedure are slightly different, the original coordinates were also subjected to three cycles of DFT-based geometry optimisation (DOI: 10.14469/hpc/9983) to better reflect the electron density in the molecule.
- The resulting wavefunctions in the form of an .fchk file (for both unoptimised and partially optimised geometries) were then used to compute a grid of total electron density points
- The density, in the form of a cube of points, was fed to Jmol using the commands
load biotin_den.cub; isosurface parameters [0.5 1 0.0005 0.05 0.95 1.00] NCI ""; color isosurface "bgyor" range -0.04 0.04;
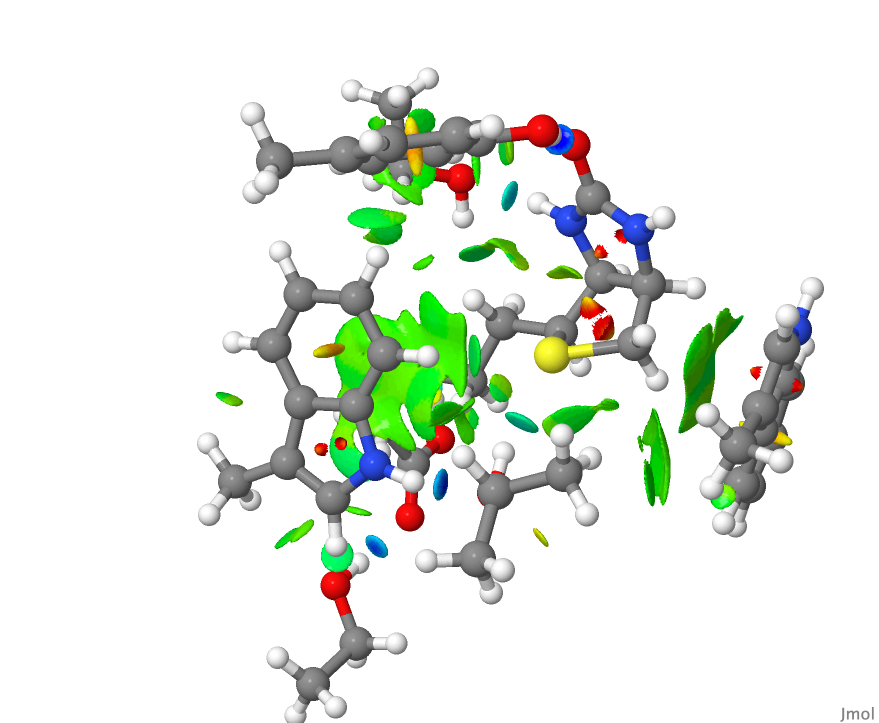
and the resulting NCI surface was written out using the command write biotin.jvxl for inclusion here. - This is the NCI plot obtained from the raw coordinates from the PDB file.
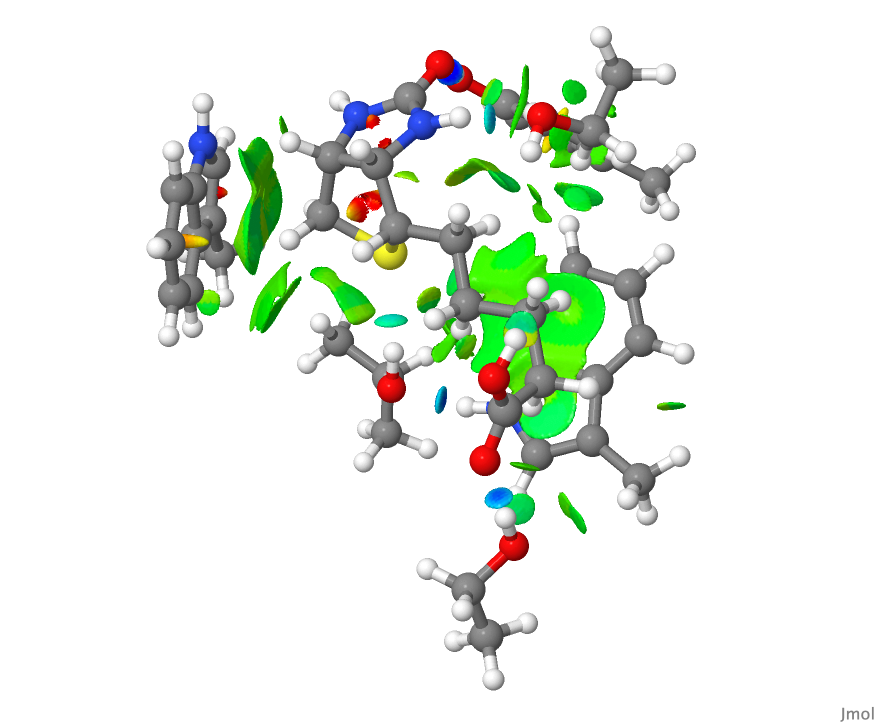
- This is the NCI plot obtained from the coordinates from the PDB file after three geometry optimisation cycles. Can you spot any differences?
- These models are now available for you to explore by clicking on the images above.
- Blue regions represent “strong” or classical hydrogen bonds. There are four of these in the NCI diagrams above and they are all compact, another characteristic of strong hydrogen bonds.
- The hydrogen bond to sulfur is somewhat weaker, and appears in the display as a compact, albeit now cyan-coloured surface.
- The remaining regions are both diffuse and green and represent weaker “interactions”. They are less compact than the classical hydrogen bonds. They do not represent a bond so much as an attractive region in the molecule and hence the term non-classical. Most are CH groups close to the π-surface of an aromatic ring, but some are also CH…O interactions.
Do go ahead and load the 3D surface. You should particularly explore the CH-π regions and note that they are not necessarily associated with a particular CH bond, but with several of these combining to form an interaction with an aromatic π region.
What might emerge is the realisation that binding interactions are not always between specific atoms as in classical hydrogen “bonds”, but also constitute “stabilising regions” between the ligand and the protein. You will probably spot several of these regions that are not actually listed in the article itself.[1] I suggest that we do not refer to CH…π bonds such as in the quoted title of this post but instead as CH…π regions.
It would be great if the entire complex could be subjected to an NCI analysis. Wavefunctions for >2000 atoms can be obtained nowadays, but it would require a bit of work to ensure the density can be computed accurately enough and at high enough cubic resolution to be useful in the context of NCI analysis.
This blog has DOI: 10.14469/hpc/9984
Author
References
- D.B. McConnell, "Biotin’s Lessons in Drug Design", Journal of Medicinal Chemistry, vol. 64, pp. 16319-16327, 2021. https://doi.org/10.1021/acs.jmedchem.1c00975
- I. Le Trong, Z. Wang, D.E. Hyre, T.P. Lybrand, P.S. Stayton, and R.E. Stenkamp, "Streptavidin and its biotin complex at atomic resolution", Acta Crystallographica Section D Biological Crystallography, vol. 67, pp. 813-821, 2011. https://doi.org/10.1107/s0907444911027806