From the last few posts here, you might have noticed much discussion about how the element carbon might sustain a quadruple bond. The original post on this topic from some years ago showed the molecular orbitals of the species CN+, which included two bonding π-types and a low lying nodeless bonding σ-orbital, all with double occupancies and adding up to a triple bond. Discussing now C2 itself, there are two remaining orbitals for consideration which we will for the purpose here call the highest occupied σ-MO or HOσMO (Σu) and the lowest unoccupied σ-MO or LUσMO (Σg) and which are more mysterious.
The HOσMO itself has one node (the lowest unoccupied or LUσMO has a further second node) bisecting the centre of the C-C bond, which makes it anti-bonding. This is emphasised by squaring the orbital (below), which shows a clear void of electron density in the C-C region. For this reason, many text books illustrating the main group diatomic molecules represent C2 with two bonds: 3-1 = 2.
| CASSCF(8,6) “HOMO”/”LUMO” orbitals and densities of C2 | |
|---|---|
| (HOσMO)@0.02au | (LUσMO)@0.02au |
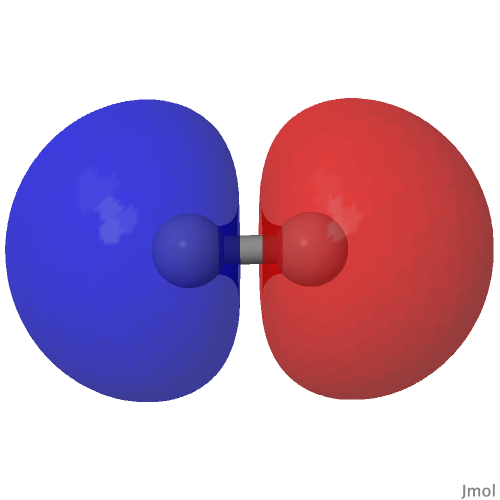 |
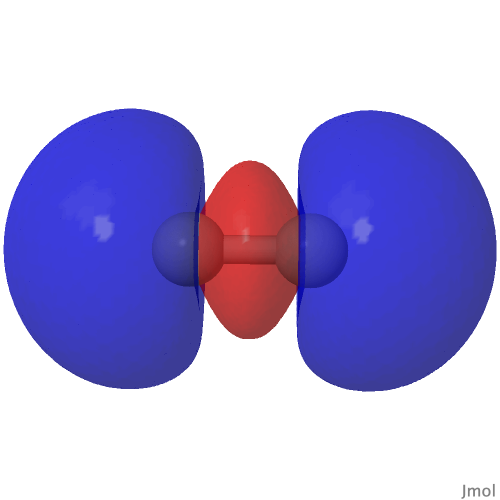 |
| (HOσMO2)@0.0004au | (LUσMO2)@0.0004au |
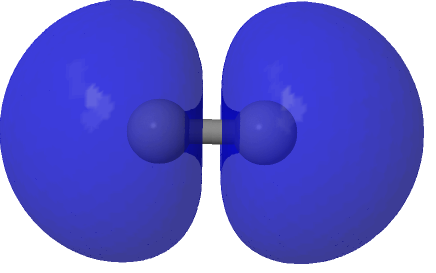 |
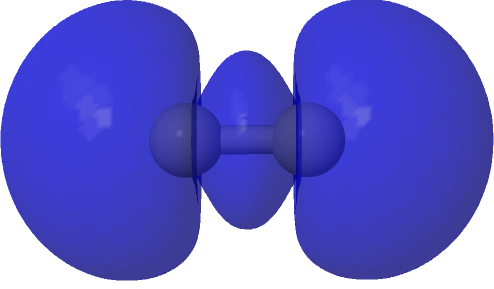 |
Now to a MCSCF(8,6)/Def2-SVPD calculation (FAIR DOI: 10.14469/hpc/8307) which means a multi-configuration calculation. Regular e.g. DFT methods assume only a single electronic configuration in which one set of doubly-occupied orbitals is variationally optimized. Thankfully, for the vast majority of molecules, this is actually a very good approximation. However for some species, and C2 is one such, this is no longer true. A CASSCF(8,6) calculation uses the 105 different electronic configurations generated by using eight electrons in an active space of six orbitals and variationally optimises them all for a self-consistent-field. The orbitals corresponding to the erstwhile single-configurational HOσMO and LUσMO are the ones shown above. Their squares are shown underneath, the latter as noted above relating to the electron density distribution in the molecule.
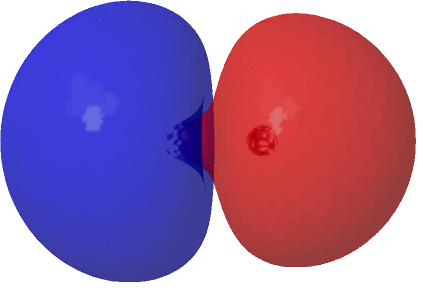
The MCSCF calculation for C2 shows that primarily two different electronic configurations contribute significantly to the total wavefunction, the one with two electrons in the original HOσMO (now of course a misnomer) having a weight of 1.573e and the configuration with two electrons promoted to the now similarly misnamed LUσMO orbital by virtue of having a weight of 0.427e.‡ This shows that this final 2e really must be described by two electronic configurations rather than one (and which reminds that the terms HOσMO and LUσMO really only apply to single-configuration methods). What difference does that make to the picture? The scaled linear combination of the two orbitals deriving from the two dominant electronic configurations for C2 below shows that each now has an “extrusion” of the original orbital creeping along the C-C axis.
| (1.573*HOσMO + 0.427*LUσMO)@0.02au | (1.573*HOσMO – 0.427*LUσMO)@0.02au |
|---|---|
 |
 |
Squaring and weighted adding shows us what is happening to the electron density. That void in the density along the C-C axis apparent in the HOσMO above has now been nicely filled with density from those extrusions deriving from the partially occupied “LUσMO”. As a result, the node in the density along the bond has now vanished. By elevating 0.427 of the electrons from the original anti-bonding HOσMO into the complementary LUσMO, a new weakly-bonding “baby” orbital with ~two-electron occupancy replaces the original antibonding HOσMO. There is however relatively little additional density placed into the C-C region because of only 0.427e transfer into the “LUσMO”. The weak bonding character also matches the “bond dissociation energy” of this fourth bond of ~17 kcal/mol as inferred by experimental measurement of the energies of the two reactions HC≡CH → HC≡C• + H•; HC≡C• → CC + H•.
| (1.573*(HOσMO)2 + 0.427*(HOσMO)2)@0.0004au |
|---|
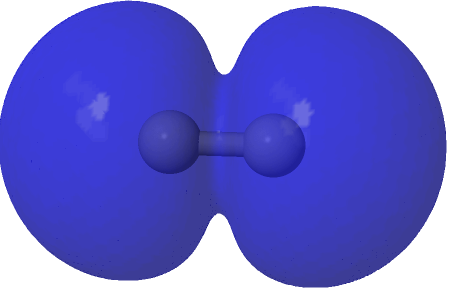 |
| The (Σg)2@0.0004au conventional σ-bond for comparison |
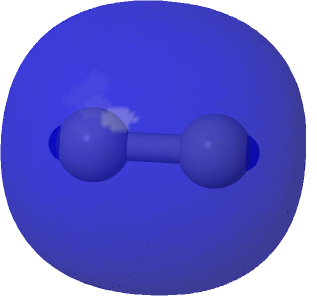 |
| (Σg)2 – (1.573*(HOσMO)2 + 0.427*(HOσMO)2)@0.0004au |
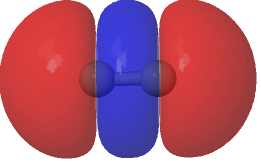 |
So by combining the appropriate occupancies of the HOσMO and LUσMO in a multi-configurational approach to C2, a new weak bond emerges, which when added to the three existing bonds referred to above gives a representation of four rather than two bonds for this molecule.
The DOI of this post is https://doi.org/gf9s
‡The weights for a CASSCF(12,12) calculation with 427350 configurations are 1.568 and 0.426. Conversely, a CASSCF(8,5) calculation on 15 configurations yields 1.533 and 0.467. The optimised geometries also show an interesting trend. Thus 8,5 = 1.198Å, 8,6 = 1.230 and 12,12 = 1.262Å. As the active space decreases, so the weight of the configuration with a populated Σg orbital increases by “concentration” into this orbital and hence the C-C bond length also decreases as the amount of density injected into the C-C region increases.