In the previous post, I showed the geometries of three large cyclic porphyrins, as part of an article[1] on exploring the aromaticity of large 4n+2 cyclic rings. One of them had been induced into a “figure-eight” or lemniscular conformation, as shown below.
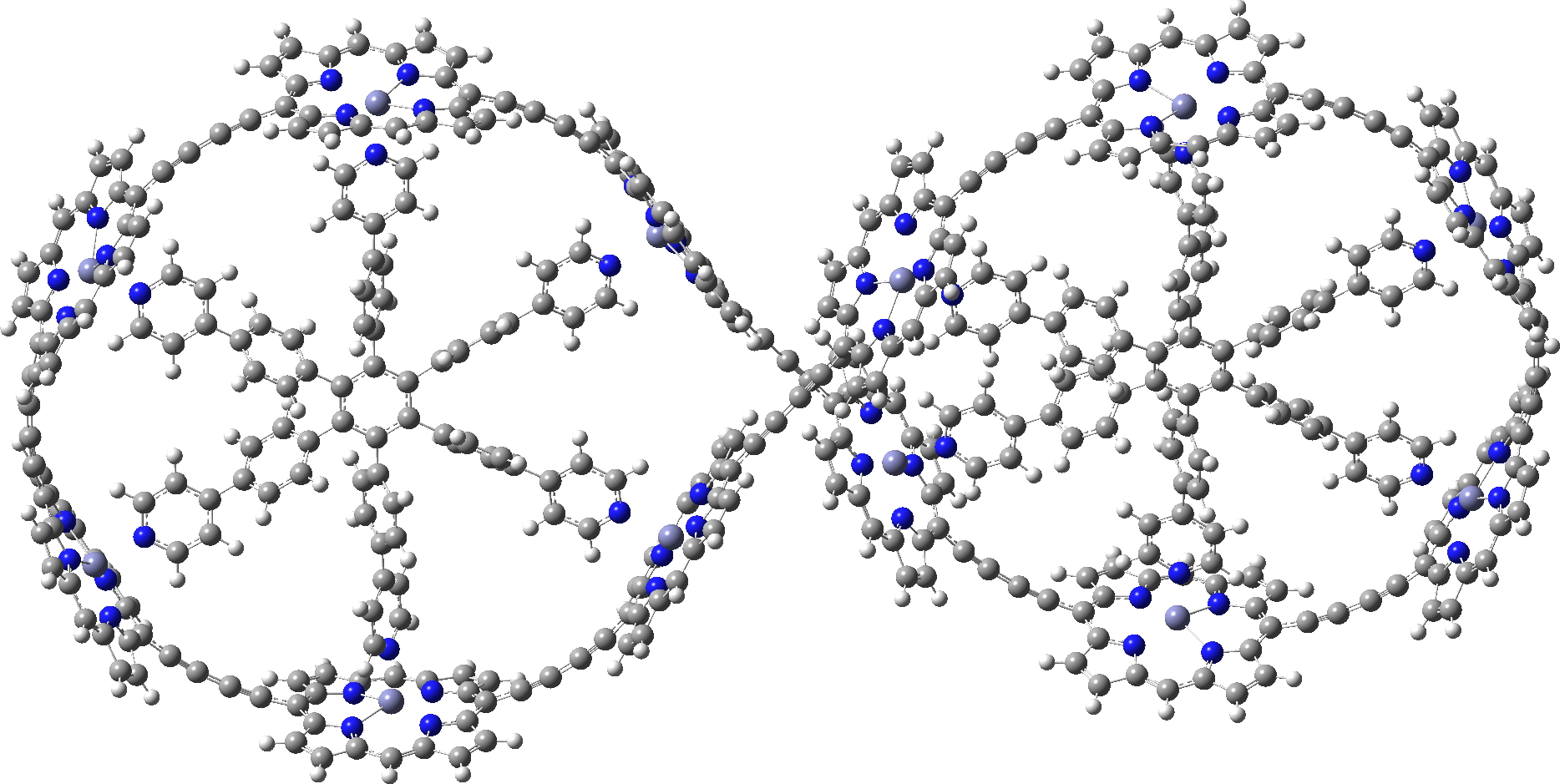
c-P12[b12]_T6f optimised with LC-ωhPBE
c-P12[b12]_T6f optimised with PM6
Any initial inspection of the geometries of these systems suggests they have a high level of symmetry, and the molecule above does have the potential for D2 symmetry, typical of such leminscates.[2] The coordinates provided as part of the article however[1] had no symmetry and in the previous post, I asked myself if the coordinates could be symmetrised. That proved possible for the first two molecules shown, and here I ask myself it it can be done for the molecule shown above. My method for symmetrising was to use the PM7 semi-empirical method.[3] For the first time in the PM-series, this latest extension includes a Grimme-style dispersion attraction correction, one that grows in magnitude with the size of the molecule. Since this is a large molecule (720) atoms, dispersion might well have an important role to play. The coordinates provided[1] were obtained using the LC-ωhPBE/6-31G* method, a functional which in its latest form does not include a built-in dispersion term, although one can be added to the earlier LC-ωPBE version of the functional. We may presume that the calculation used to obtain the coordinates shown above does not include such a dispersion term.
Here is the PM7 optimised version. Firstly, it has C2-symmetry only and not the higher D2. The reason is that the four porphyrin rings at the point of the ring crossover, which in the original version show no ring-ring stacking, now indeed show stacking of a pair of porphyrin rings when dispersion is included in the calculation.
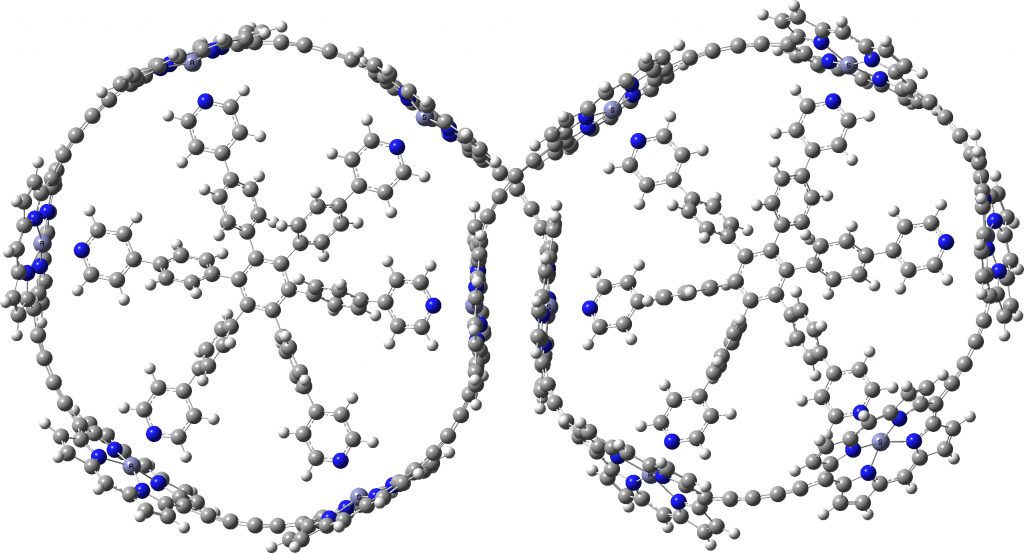
c-P12b12_T6f optimized with PM7
Whether such stacking, which does significantly perturb the overall geometry has any impact on the inferred aromaticity, remains to be established. But it reinforces the conclusion that when dealing with large molecules, it is absolutely essential to include a good quality dispersion correction, otherwise the geometries so obtained may differ significantly from reality.
Author
References
- M. Rickhaus, M. Jirasek, L. Tejerina, H. Gotfredsen, M.D. Peeks, R. Haver, H. Jiang, T.D.W. Claridge, and H.L. Anderson, "Global aromaticity at the nanoscale", Nature Chemistry, vol. 12, pp. 236-241, 2020. https://doi.org/10.1038/s41557-019-0398-3
- H.S. Rzepa, "A Double-Twist Möbius-Aromatic Conformation of [14]Annulene", Organic Letters, vol. 7, pp. 4637-4639, 2005. https://doi.org/10.1021/ol0518333
- J.J.P. Stewart, "Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters", Journal of Molecular Modeling, vol. 19, pp. 1-32, 2012. https://doi.org/10.1007/s00894-012-1667-x
Dear Prof Rzepa,
I am happy that you find these molecules attractive. The attraction between two porphyrins and its stacking seems to me as a bit surprising perturbation of the structure. It bends the butadiyne linkers significantly. Isn’t it possible that semi-empirical PM7 overestimates the gain from having a flat conjugated system and underestimates the bending energy of the butadiyne? Further problem might be that the molecules were studied oxidised to 6+ – 12+ state and the charge delocalisation in such scaffold will be even less accurately described by semiempirical methods. Or would you disagree?