Here is another molecule of the year, on a topic close to my heart, the catenane systems 1 and the trefoil knot 2[1] Such topology is closely inter-twinned with three dimensions (literally) and I always find that the flat pages of a journal are simply insufficient to do them justice. So I set about finding the 3D coordinates.
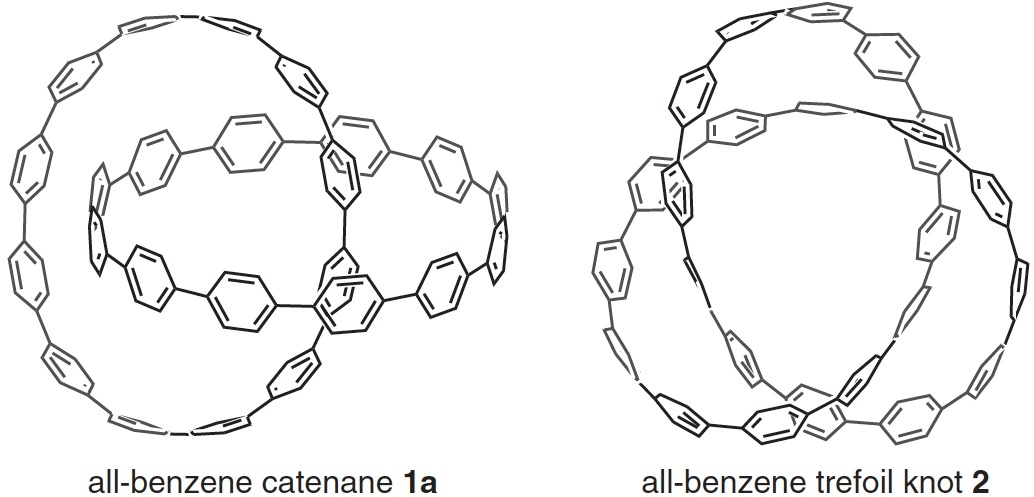
The most obvious place to start is the supporting information. I show below a little snippet of what I found, which is fairly typical of such data in the PDF-based SI documents accompanying most articles.
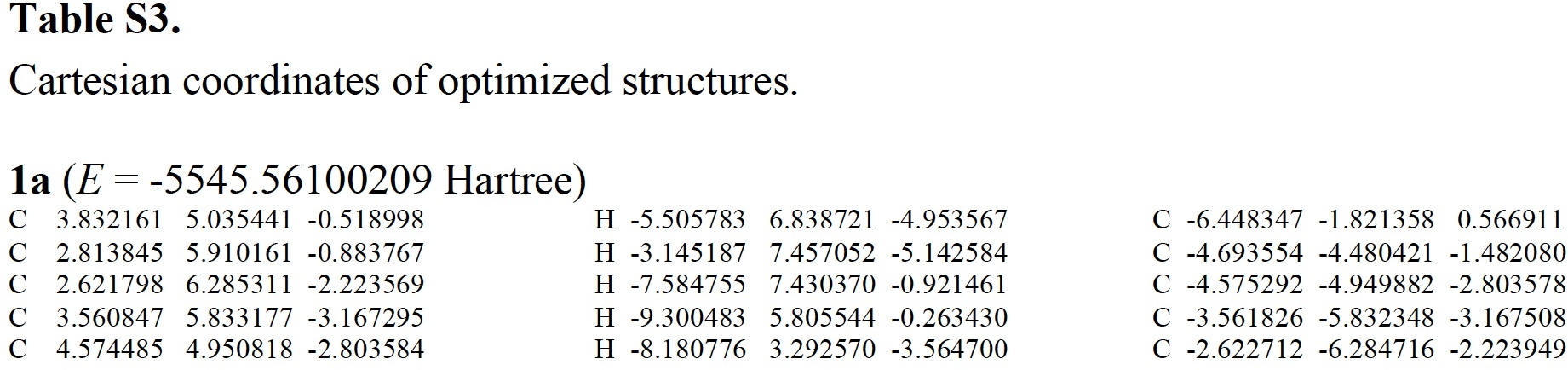
A bit of knowledgeable text-editing is needed to convert these into something that can be displayed as a rotatable 3D model.‡ For this example, the three-column mode did not actually prove too problematic (but sometimes you have to work very hard to reduce it to the single column mode required for coordinates) and one has to remember to notice and remove the pagination text from the coordinates. Here I include the structure as a static 2D image which when clicked expands to a 3D model. Sadly, I know of no journal that offers up this relatively simple service as part of its “added-value” to the publication processes; it has been possible to do this since 1994![2],[3] I also found that the provided coordinates could be symmetrised to D2.
1a, which are here symmetrised to D2 symmetry.
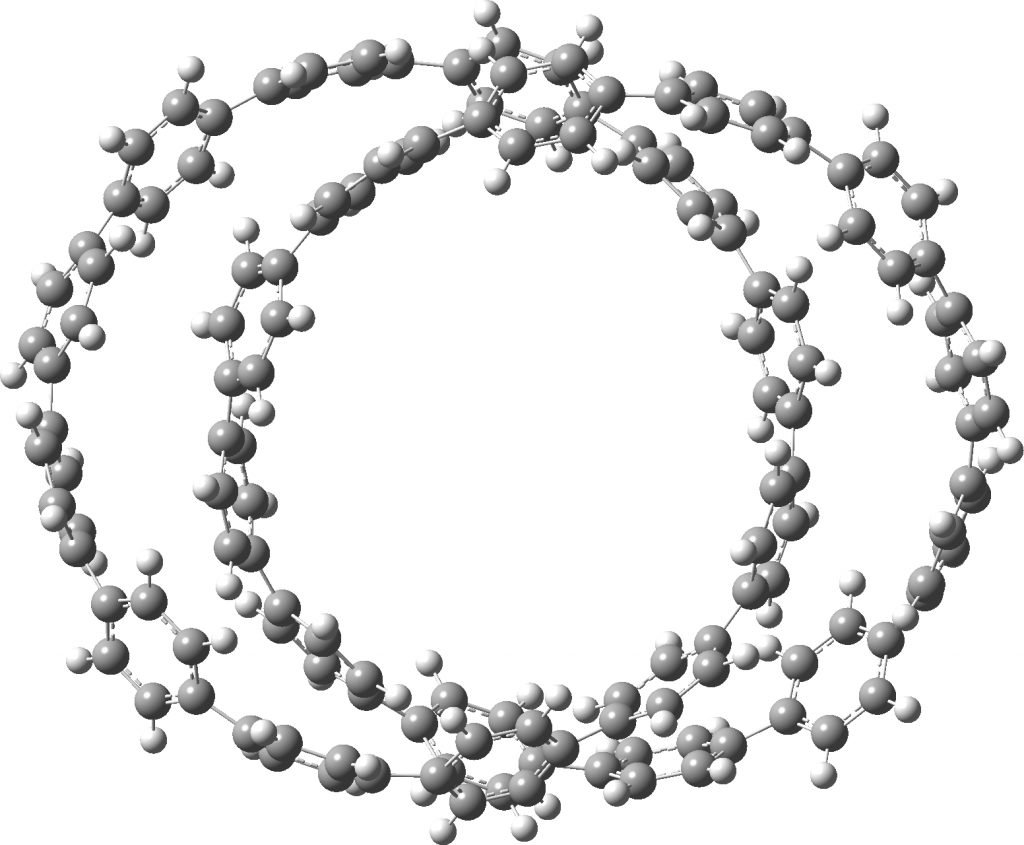
Molecule 2 posed a new challenge. The coordinates when extracted from the SI had 480 atoms, double that of 1a. When displayed they overlayed each other. Clearly two sets of 240 atoms was the answer (the first probably in error) only the second set of which displays as a trefoil knot. The coordinates here can be symmetrised to D3, as appropriate for a trefoil knot.
2, which here are symmetrised to D3 symmetry
The above coordinates are computed using quantum mechanics at the B3LYP-D3/6-31G(d) level. What about the crystallographic coordinates? Here again a little expertise is needed to obtain these.
- Just after the article acknowledgements, the CCDC identifiers are given as 1860595-7 and 1908693. These values are resolved using www.ccdc.cam.ac.uk/structures/ which allows the download of a CIF file.
- These files reveal that a number of solvent and other molecules have been occluded into the structures, and for clarity it helps to edit all these out as well as disordered atoms with partial occupancy.
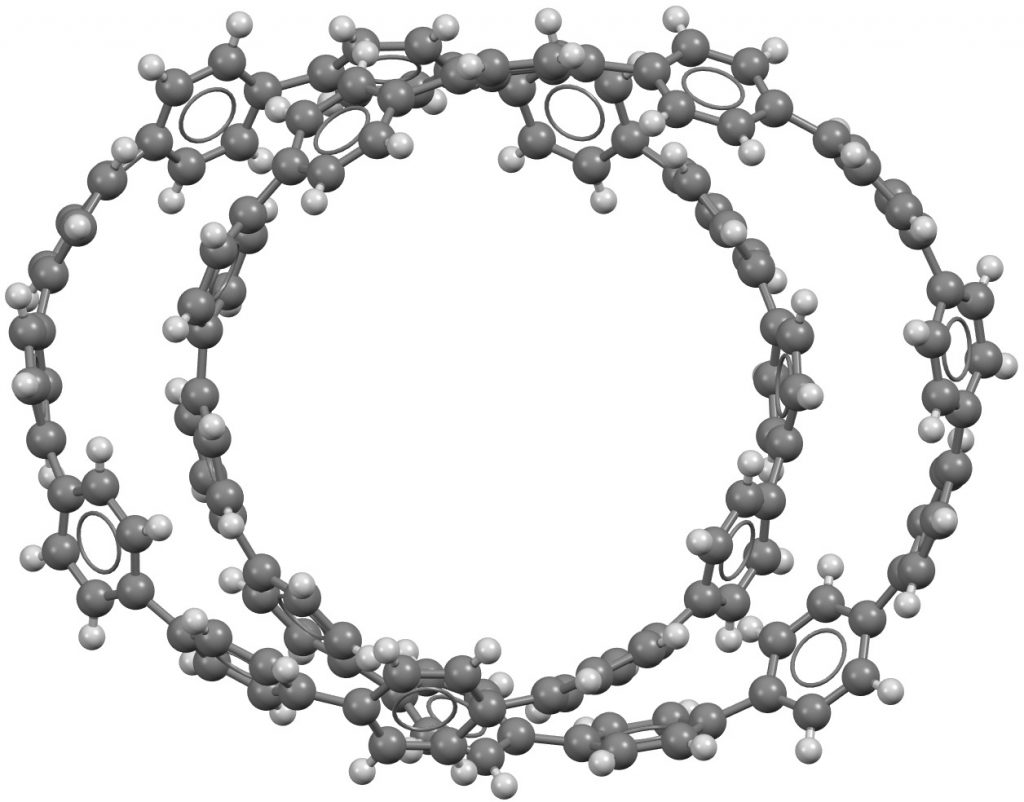
Crystallographic coordinates for 1a. DOI: 10.5517/ccdc.csd.cc20g36t
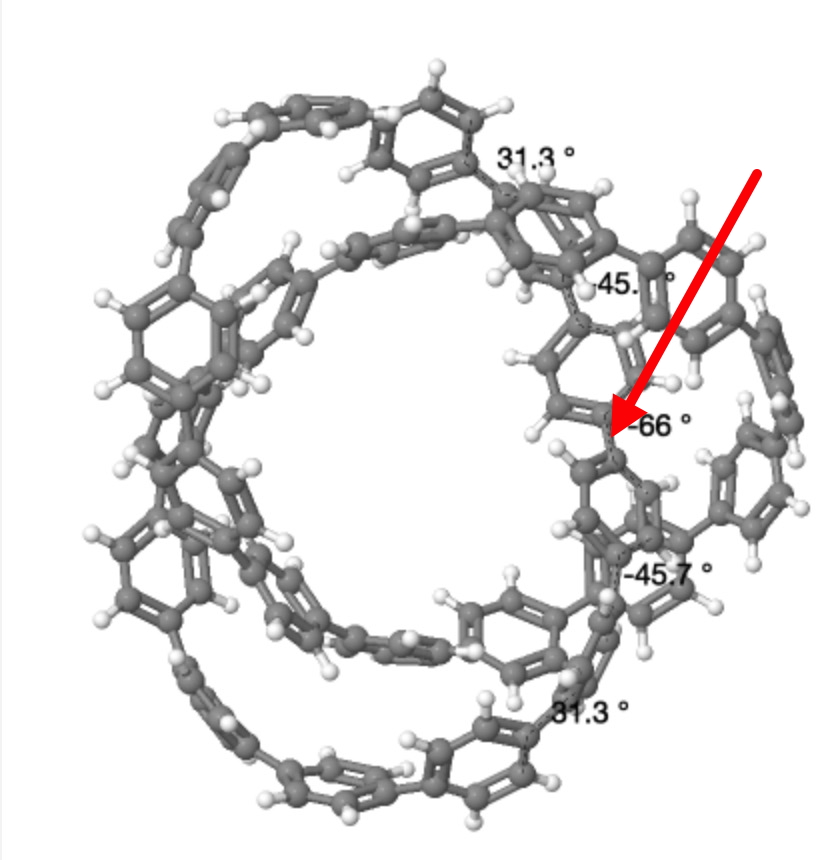
Crystallographic coordinates for 2. DOI: 10.5517/ccdc.csd.cc20g37v
Each of these experimental structures is also allocated a DOI of its own, which can be accessed from the captions above, if you want to view the un-edited coordinates. I have also made available my coordinates produced for the display here as a FAIR dataset (DOI: 10.14469/hpc/6470), together with metadata such as the InChI descriptors to improve its discoverability.
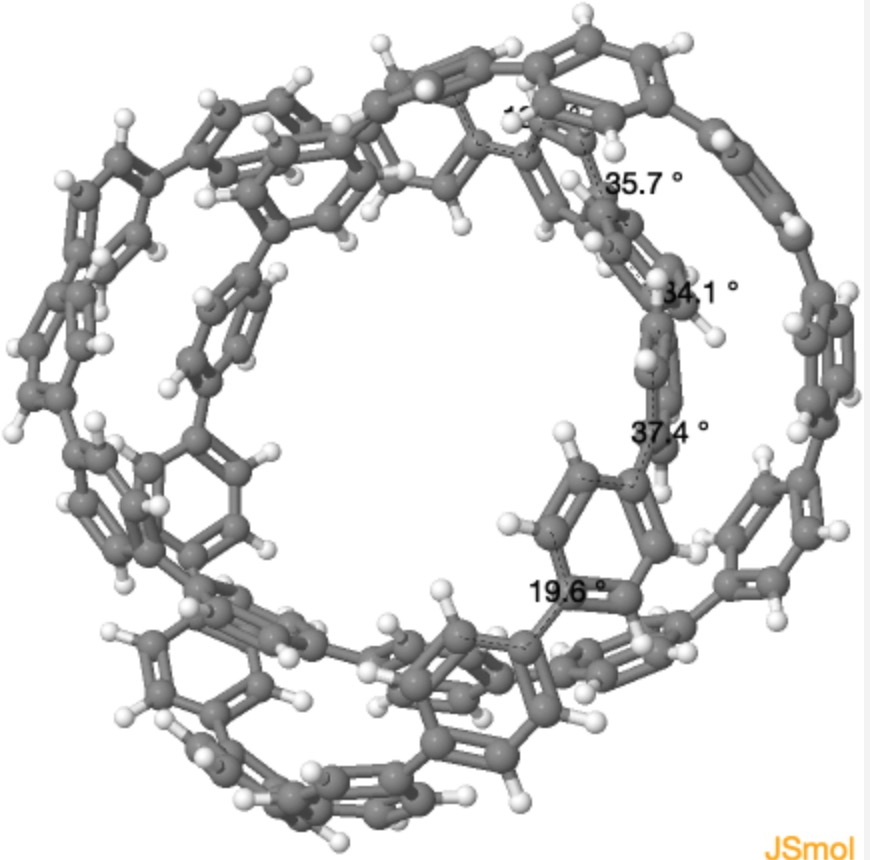
Having acquired and displayed both the calculated and the measured coordinates, I noticed one oddity. The calculated structure for 2 symmetrises to D3 symmetry, but the measured crystal structure only to C2 symmetry. This is due to a “kink” in the twists for the latter coordinates, a single region where the Ar-Ar single bond is twisted by 66°. This kink is absent in the calculated coordinates, where the largest dihedral angle at the Ar-Ar bond is only 37°. Is this effect real? What does it tell us about the conjugations and extended aromaticity of this system? Such twist localisation has been previously noticed in cyclacenes.[4] I think this observation highlights the need to have readily accessible 3D structures of such novel systems, if only to allow readers to spot such apparently anomalies.
So, with a little bit of knowledge and effort, one can indeed proceed from a published article to viewing aspects of the three-dimensional topology of the molecules discussed. I just feel it would be good if these aspects could be better integrated into the article itself, since I suspect that the additional effort and knowledge required to go further is probably not going to appeal to most readers.‡
‡Tidying up the PDF cartesian coordinates into a list of atomic number and a set of three coordinates per line of text is relatively simple. To coerce this format into a visualisation program takes more knowledge. Direct conversion to eg a standard molfile is not possible. I instead add a header to the coordinates to make it suitable for visualisation using the Gaussview program. Another good program for handling this is wxMacMolPlot which supports the veritable Xmol XYZ format, but again a correct header at the top of the file is needed for this program to recognise the file. As I noted, only a knowledgeable user would be able to do this, and the average reader is unlikely to go down this road.
Author
References
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota, and K. Itami, "Topological molecular nanocarbons: All-benzene catenane and trefoil knot", Science, vol. 365, pp. 272-276, 2019. https://doi.org/10.1126/science.aav5021
- H.S. Rzepa, B.J. Whitaker, and M.J. Winter, "Chemical applications of the World-Wide-Web system", Journal of the Chemical Society, Chemical Communications, pp. 1907, 1994. https://doi.org/10.1039/c39940001907
- O. Casher, G.K. Chandramohan, M.J. Hargreaves, C. Leach, P. Murray-Rust, H.S. Rzepa, R. Sayle, and B.J. Whitaker, "Hyperactive molecules and the World-Wide-Web information system", Journal of the Chemical Society, Perkin Transactions 2, pp. 7, 1995. https://doi.org/10.1039/p29950000007
- S. Martín-Santamaría, and H.S. Rzepa, "Twist localisation in single, double and triple twisted Möbius cyclacenes†", Journal of the Chemical Society, Perkin Transactions 2, pp. 2378-2381, 2000. https://doi.org/10.1039/b005560n
Dear Henry, just a comment to your footnote:
The easiest way to get the coordinates into a viewer is to use the XYZ format. It just involves copying the text from the pdf into a plain text editor, removing blank lines and adding 2 header lines: first one with the number of atoms, second line empty or with a comment. That is read by Jmol.
Angel,
Yes, that is pretty much what I try to do. You still have to remove pagination marks, and then add a header to the coordinates. You have to know this (it is not described for you in the SI often). But even the simple “copying the text from the pdf into a simple text editor” can hide problems. If that text is double column, you may have a problem. Sometimes the end-of-line markers are not recognised in your “simple text editor” and you have to break the lines yourself. Once, the PDF was from a Chinese source, and the underlying font family did not map to actual numbers; the numbers in the PDF emerged as non-numerical characters in the “simple text editor”. I have encountered many different types of issue with this apparently simple operation over the years. It should not have to be so!