In the pipeline reports on an intriguing new ring system acting as an isostere for morpholine. I was interested in how the conformation of this ring system might be rationalised electronically and so I delved into the article.[1] Here I recount what I found.
The basis for the isosteric claim can be found in the conformational analysis reported in Figure 4.‡ The N-diazine ring in A is found to be co-planar with the morpholine ring as shown in the diagram (the dihedral measured is indicated using boldened bonds). Compound D contains the cyclopropanated variation, which is postulated as isosteric at least in part on the basis that it too is co-planar, with a dihedral angle of ~170° (with a second slightly higher minimum having a value of ~10°) and hence might be capable of acting as an isostere to the morpholine.
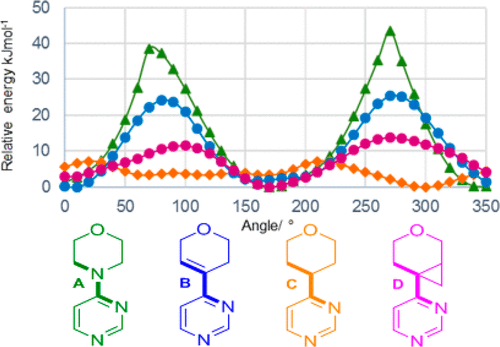
Figure 4. Dihedral scanning plots for various pyrimidine fragments using DFT/6-31G**.
I was intrigued as to why a saturated sp3-carbon would exhibit the same behaviour as a nitrogen centre. The latter has a lone pair oriented at 90° to the aryl ring, the resulting conjugation favouring co-planarity. But how would that sp3-carbon centre do the same? Time to do some calculations, and hence on to the supporting information (SI) for the article in an effort to get a starting base – initially to replicate the calculation results shown above. I start by focusing on the value quoted above, ~170°. Note the ~, since I obtained that visually from the figure. In fact it must remain “~“, since no further geometrical information is available from the SI. Quickly I also realized that replication must also remain elusive, since the caption to the figure is the only information on the calculations which were used to produce figure 4. DFT you see is a generic term, standing for density functional theory. But in that theory the functional has to be defined; there are possibly about 500 different functionals that have been used in the literature. We do get a citation to the method (ref 25 in the article) which is to the commercial Jaguar program system. Herein lies a problem. Programs implement what might be described as default calculation options and quite possibly it is the default option that has been invoked here. A licensed user of Jaguar can probably find out what that default option is and hence can expand DFT to the actual functional used. But unfortunately I am not a licensed user, and even if the default option could be tracked down to an online manual somewhere, there is no certainty it was actually used to produce Figure 4.
So here I make my first plea. The SI for this article is not fully FAIR! In this instance it contains no accessible data that can be used to replicate the results reported. At a minimum, if DFT based results are going to be reported, then FAIR data containing the input(s) used for the calculation and one or more outputs should be made available. Perhaps then if one is lucky, those outputs might declare any default assumptions, such as the precise DFT method used.†
I therefore went ahead with my own calculations, deciding to use B3LYP (being my declared DFT functional) with the 6-311++G(d,p) basis (an improvement on the 6-31G** (≡ 6-31G(d,p) basis set declared as used for Figure 4). I did two variations, one without a D3+BJ dispersion attraction correction and one with. It is now recognised that such corrections can be important, even for small molecules. Because we do not know the nature of the DFT method used in the article itself, we do not know if it incorporates such corrections or not. The results are shown below, with a FAIR data location of DOI: 10.14469/hpc/5990♥

The two minima from the new B3LYP+D3BJ/6-311++G(d,p) calculation have dihedral values of -139.4° and +55.6° with dispersion included and essentially the same without, indicating that dispersion has only a small effect on the conformational geometry (the top trace above is without dispersion). These values are different from the ones inferred from Figure 4, being closer to gauche than to co-planar. These new values can be rationalised as allowing good overlap between a C-C bond of the cyclopropane and the π-system of the aromatic ring (dihedrals 75 and 88° for the two minima vs 90° for the overlap of the N-lone pair). The values for the conformation implied in Figure 4 are 41 and 55°, which is less favourable hyperconjugative overlap. The rotational barriers are ~18 and 25 kJ/mol, rather higher than those obtained visually from Figure 4, but still indicating a relatively flexible molecule which can probably adopt a relatively low energy co-planar isosteric conformation in the correct environment.
There is however some more information about these molecules reported in the article,[1] being a small molecule crystal structure for a related compound 12b (quoted for Figure 5 as CCDC 1864315). To quote, “the small molecule crystal structure of 12b confirms coplanarity in the solid phase”. However, the dihedral angle for this crystal structure is not given either in the text or the SI. A search of the CCDC database reveals no entries in May 2019 database (the data is clearly too new to have been indexed there) and unfortunately the article SI contains no atom coordinates.♣♣ The calculations reported in Figure 4 and the ones in the plot above are of course for an isolated molecule. Once I manage to acquire the crystal coordinates, it should be possible to see if there are any intermolecular interactions which are a factor in explaining why the geometries of the isolated molecule and its crystal form might differ in co-planarity.

Until then I conclude that the inclusion of FAIR data pertaining to this co-planarity in the article itself would certainly have helped to resolve the origins of the difference in the geometries reported in the article and my own calculations reported here; it may still be of course that functionals other than B3LYP+D3BJ reproduce the crystal structure better. Nonetheless, I think there is a more rational electronic basis for the conformation of the N-aryl ring in the isolated molecule based on the dihedral angles reported here, whilst an attempt to replicate the values reported in the article itself[1] based on further information would also be useful.
‡Reprinted with permission from [1]. Copyright 2019 American Chemical Society.
†In this article[2], making quite some waves, you can find a fascinating discussion of the perils of using “packaged” programs in which many “defaults” are allowed to persist by the user. In this particular case, the default was the size of the integration grid in the DFT calculation. This article make the very alarming case that for many years the default size in at least one popular DFT program was not good enough to ensure that resulting calculated free energies were sufficiently accurate to sustain many conclusions for regio and stereoselectivity out there in the wild. A awful lot of computational chemistry derived results might be wrong! You may only be slightly re-assured that the default grid sizes used for the calculations reported in this blog, at least for the last five years or so, are suitably larger than the one critiqued in this article.
♥From which you find a keyword integral=(acc2e=14,grid=ultrafine) defined, which ensures that not only is the integration grid declared, but also that the integral accuracy is pumped up beyond the program defaults of 12. We have found that this is very often helpful for calculation of frequencies.
♣ New entries, not yet available in the distributed database, can be accessed as e.g. https://www.ccdc.cam.ac.uk/structures/search?pid=ccdc:1864315 The dihedral is 167.7° for one conformation of compound 12b. ♣This has now also been assigned DOI: 10.5517/ccdc.csd.cc20kz6s The coordinates obtained from this source correspond to an absolute stereochemistry of 1R,6S, or 12a in the article.
Author
References
- H. Hobbs, G. Bravi, I. Campbell, M. Convery, H. Davies, G. Inglis, S. Pal, S. Peace, J. Redmond, and D. Summers, "Discovery of 3-Oxabicyclo[4.1.0]heptane, a Non-nitrogen Containing Morpholine Isostere, and Its Application in Novel Inhibitors of the PI3K-AKT-mTOR Pathway", Journal of Medicinal Chemistry, vol. 62, pp. 6972-6984, 2019. https://doi.org/10.1021/acs.jmedchem.9b00348
- A.N. Bootsma, and S. Wheeler, "Popular Integration Grids Can Result in Large Errors in DFT-Computed Free Energies", 2019. https://doi.org/10.26434/chemrxiv.8864204.v1
There is some additional information provided in the article: The absolute stereochemistry of 12a and 12b are inferred from the small molecule crystal structure of 12b (Figure 5b, see Supporting Information),36 which has negative optical rotation. For 12b (1S,6R), three possible conformations (there are more) were investigated here for their calculated optical rotations using M062X/6-311++G(d,p)/SCRF=chloroform, giving [α]D rotations of +118°, +217° and -54°. The enantiomer 12a (1R,6S) would of course have rotations of the opposite sign. See 10.14469/hpc/5990 for the data.
The article reports [α]D of -100 for 12a and +70 for 12b. The lowest energy of the three conformations noted above has [α]D +118, which matches 12b.
Hello, thanks for this, very interesting. Most in silico chemists I have worked with, in industry and academia, do not seem to keep clear “lab books” containing procedures and parameters used for calculation. I am not sure why this is. A synthetic chemist does not get away, usually, with just saying “we did a suzuki reaction” – conditions are important – but you often see “docking was use” or “DFT calculations showed” etc…. Anyway, the CSD structure is now available – from a quick look appears to be maybe 10 degrees from coplanar. Would be interested to hear your thoughts.
…this may as you suggest be due to solid packing as the group in question stacks partly over an aromatic of another molecule
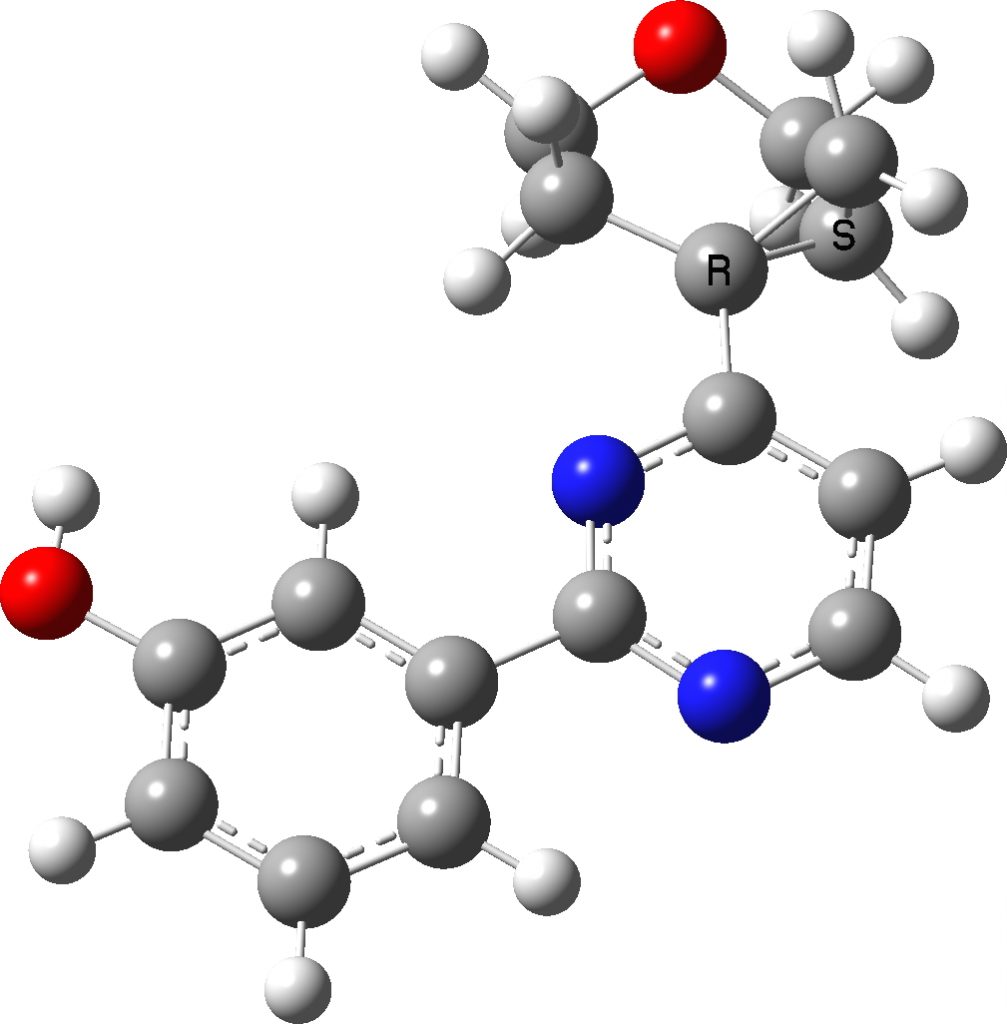
Here is the computed (B3LYP+GD3+BJ/6-311++G(d,p)) structure. The dihedral angle is computed as 173.7°, very similar to the crystal structure. From which we conclude that the dihedral may be significantly influenced by the nature of the N-aryl group, and that D as a model may not be perfect. It also suggests that the basic method used here is tested at least once as reasonably reliable.
I noted above that the calculated optical rotation [α]D for 12b varied according to the conformer studied. Here I show another chiroptical measurement that can be made in solution, the vibrational circular dichroism spectrum. Sometimes such spectra can be less vulnerable to conformational orientations. Here the results show that the VCD also changes, but that several vibrations could be used as markers identifying conformations.
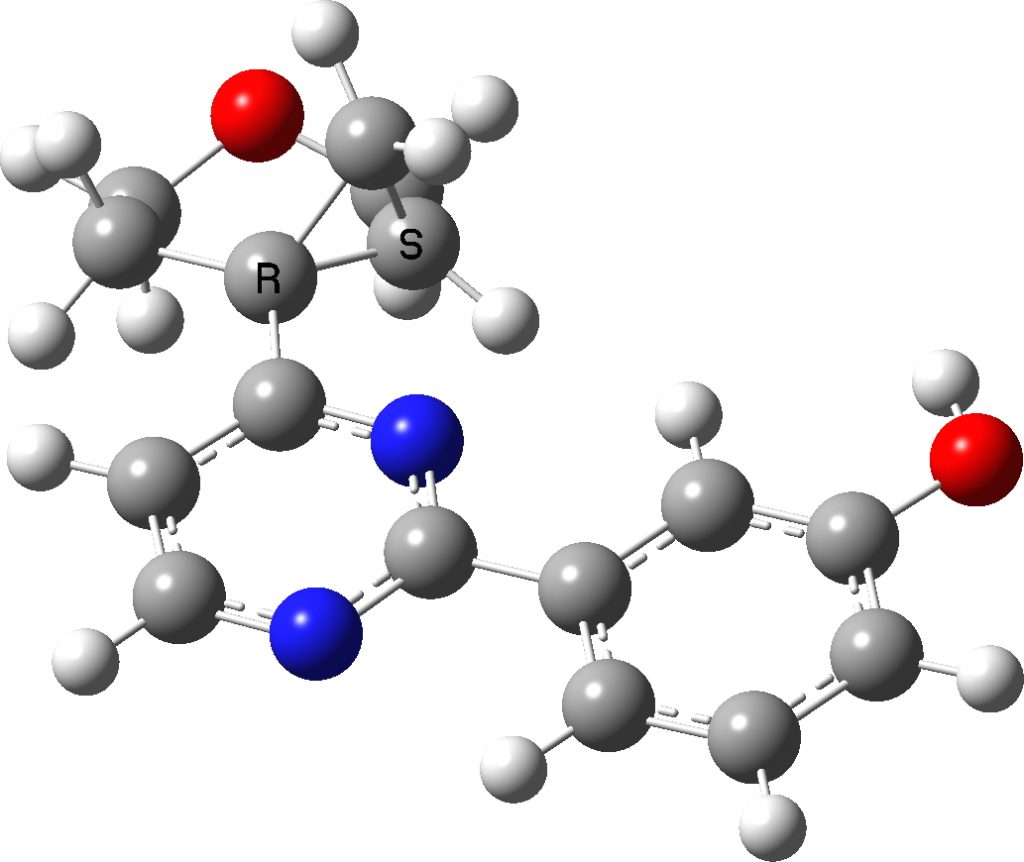
Rotamer 1
Rotamer 2