Here is the concluding part of my exploration of a recently published laboratory experiment for undergraduate students.[cite]10.1021/acs.jchemed.7b00566[/cite] I had previously outlined a possible mechanistic route, identifying TS3 (below) as the first transition state in which C-C bond formation creates two chiral centres. This is followed by a lower energy TS4 where the final stereocentre is formed, accompanied by inversion of configuration of one of the previously formed centres (red below). Now I explore what transition state calculations have to say about the absolute configurations of the final stereocentres in the carbaldehyde product.

Previously, I had clarified that using the (S)-configuration of the prolinol catalyst results in the major stereochemical isomer as (1R,2S,3S), as shown above. TS3 is now explored in more detail as four♠ stereochemical isomers, depending on the facial selectivity of the two nominal double bonds used to create the new C-C bond (dashed line above). The subsequent step TS4 is of lower energy and hence is not rate determining in a classical sense at least. It involves inversion of configuration to eliminate the chlorine to form the second C-C bond. Then a last tidying up step where the imine is hydrolysed down to the carbaldehyde, a process in which no stereocentres are involved. The computational method used is as before, B3LYP+GD3BJ/6-311G(d,p)/SCRF=chloroform and T=273.15K. This selection is so that a good quality recent dispersion correction (GD3BJ) can be used, since dispersion attractions in large part often control stereochemical outcomes.
The results are summarised below for two models; (a) a partial model in which the products of the first steps of the reaction, namely water and amine base, are excluded; (b) a fuller model in which both water and amine base are allowed to interact with the transition state. The R’ group (Me replacing heptyl) is placed trans to the en-iminium group for the four transition states arising from facial selectivity. Two more are derived from bond rotation (blue above) to place the R’ group cis to the iminium group.
| Transition state models.♥ | |||
|---|---|---|---|
|
Stereochem of product[cite]10.1021/acs.jchemed.7b00566[/cite] |
ΔΔG273‡ Model (a) kcal/mol |
ΔΔG273‡ Model (b) kcal/mol |
TS3 (click for model) |
|
(1R,2S,3S) ≡ 4a “major” isomer |
0.0 | 0.0 | 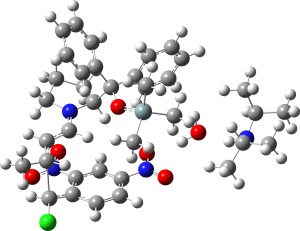 |
|
(1S,2S,3R) middle isomer? |
6.8 | 0.3 | 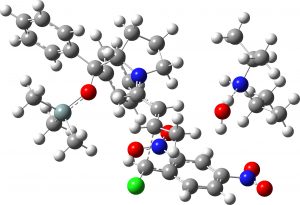 |
|
(1S,2S,3S) = 4b “middle” isomer |
7.9 | 1.8 | 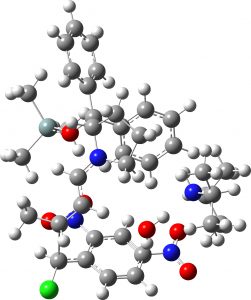 |
|
(1R,2R,3S) ≡ 4c “minor” isomer |
0.9 | 0.7 | 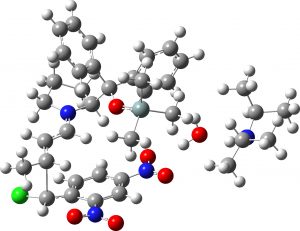 |
|
(1S,2R,3R) undetected isomer |
7.4 | 1.8 | 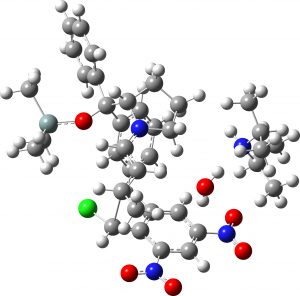 |
|
(1S,2R,3S) = 4d undetected isomer |
7.8 | 2.0 | 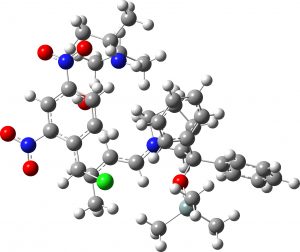 |
♥FAIR data DOI 10.14469/hpc/4704 The links to ΔΔG273‡ are to a DataCite metadata search of the free energy values for these species as described here.
The following conclusions can be drawn.
- The major 1R,2S,3S isomer resulting from use of (S)-chiral auxiliary agrees with experimental assignments in being the lowest in activation free energy for both models (a) and (b).
- Similarly the experimentally undetected 1S,2R,3R isomer 4d also has the highest activation free energy.
- There is however a mismatch between the experimental chiral assignment for the “middle” isomer and the calculations. The predicted stereochemistry deriving from the latter is derived from one of the four possibilities arising from Re/Si C=C facial selection when forming a bond between the two double bonds (TS3 = 1S,2S,3R). The experimental assignment[cite]10.1021/acs.jchemed.7b00566[/cite] implies a mechanism that involves rotation about one C=C bond (and not of a C=C face), as indicated by the blue arrow in the diagram above (= 1S,2S,3S). This is not intrinsically unlikely, since in species 6, following the first C-C bond formation, the pertinent C-C bond is now closer to single than double. This implies however that stereochemistry is determined AFTER the rate limiting transition state is passed. Incorporating such a rotation into TS3 itself for such stereochemistry (1S,2S,3S) makes the free energy slightly higher than for the unrotated TS3 (1S,2S,3R). So we might conclude that the stereochemistry observed for 4b (the “middle” isomer”) could be the result of dynamic effects such as bond rotation after the rate limiting transition state is passed. It might also be that the (1S,2S,3S) stereochemistry indicated in the article[cite]10.1021/acs.jchemed.7b00566[/cite] for 4b is mis-assigned.
- The minor isomer 1R,2R,3S ≡ 4c has a relative energy in both models (a) and (b) that matches perfectly its low abundance.
- The undetected isomer suffers from the same issue as the middle isomer. Its stereochemistry would be 1S,2R,3R from the Re/Si C=C facial selection criterion used to construct TS3, or 1S,2R,3S as shown in the article.[cite]10.1021/acs.jchemed.7b00566[/cite] Since it is undetected, there is no experimental data for comparison.
- The full model (b) appears to replicate the observed results better, in predicting three observable stereoisomers (ΔΔG‡ ≤ 1.0 kcal/mol) and one unobserved isomer (ΔΔG‡ ≥ 1.8 kcal/mol) This has important implications for such modelling, implying that incorporating species not directly involved in the bond making/breaking can nevertheless play a subtle role in the stereochemical outcomes.
The stereochemistry of the formation of two new stereogenic centres during the carbon-carbon bond formation by reaction between the ion-pair of an en-iminium cation and a benzylic anion using a chiral auxiliary has been modelled using a DFT theory which incorporates a good quality dispersion correction term. The very act of constructing such models forces one to inspect the stereochemistry very carefully, and for this purpose the CIP (Cahn-Ingold-Prelog) notation is invaluable. Two versions of such a model both agree on the nature of the major product of this reaction, and they also agree on what is likely to be an unobserved product. But one issue remains to be resolved, the nature of the second most abundant isomer, the “middle” product. Two of the three chiral centres present in the resulting cyclopropane derivative are introduced during the first C-C bond formation between the ion-pair, whilst the third results from a lower energy downstream final C-C coupling, accompanied by inversion of one of the previously introduced stereogenic centres due to elimination of chloride. Here, the experimentally assigned stereochemistry for the “middle” product would require bond rotation AFTER the first C-C bond formation. To computationally model that would probably require the molecular dynamics trajectories to be mapped out. But before doing this, it is worth flagging the need to carefully re-verify the experimental stereochemical assignments for this “middle” product.
♠Because the final product has three chiral centres, a total of eight possible stereoisomers could result, arranged as two sets of eight enantiomeric pairs depending on the chirality of the auxiliary used. The published article (caption, Figure 3) shows dihedral angles for each possible diastereomer of the cyclopropanation, accompanied by four structures. The other four isomers are enantiomers of these four. In this post, six of the eight stereoisomers possible using the S-chiral auxilliary are shown in the table above.