Consider the four reactions. The first two are taught in introductory organic chemistry as (a) a proton transfer, often abbreviated PT, from X to B (a base) and (b) a hydride transfer from X to A (an acid). The third example is taught as a hydrogen atom transfer or HAT from X to (in this example) O. Recently an article has appeared[1] citing an example of a fourth fundamental type (d), which is given the acronym cPCET which I will expand later. Here I explore this last type a bit further, in the context that X-H bond activations are currently a very active area of research.

To help understand these four types, I have colour-coded the electron pair constituting the X-H covalent bond in red.
- In mechanism (a), this electron pair stays with X, thus liberating a proton which is captured by the base.
- The hydride transfer (b) is so-called because in fact this electron pair travels together with the proton, hence the term hydride or H–.
- Hydrogen atom transfers as in (c) in effect transfer both a proton and one electron to another atom (oxygen in the example above), leaving behind one electron on X. The electron and the proton are said to travel together as a “true” hydrogen atom.
- The fourth mechanism (d) is fundamentally different from (c) in that whilst the electron and the proton travel in concert (at the same time), they do not travel together. In this example the proton travels to the oxygen, whilst the electron travels to the iron centre, in the process reducing its oxidation state. This mode is now called a concerted proton-coupled electron transfer, or cPCET as above.
The tool employed to distinguish between mechanisms (c) and (d) is the IBO or intrinsic bond orbital localisation scheme.[2] One practical advantage of such a scheme over better known localisation methods such as NBO (Natural bond orbitals) is that IBOs can be made to transform smoothly during the course of a reaction, as followed by say an IRC (Intrinsic reaction coordinate). NBOs may instead show discontinuous behaviour along a reaction IRC. Klein and Knizia have located transition states for examples of both (c) and (d) above and studied the IBOs along such IRCs. The two IBO reaction transformations are very different, as illustrated below (used, with permission, from the article itself). For the HAT type (X=C above), an α-spin IBO morphs from a C-H bond into a H-O bond, whilst the β-spin counterpart morphs from being located on the C-H bond into a carbon-centered radical. For the cPCET mode, the α-spin IBO morphs from C-H to a C-centered radical, but the β-spin region grows onto an iron d-orbital. It is in fact even more complex than the diagram above implies, since some reorganisation of the O-Fe region occurs and the H…:O region is still anti-bonding at the transition state.
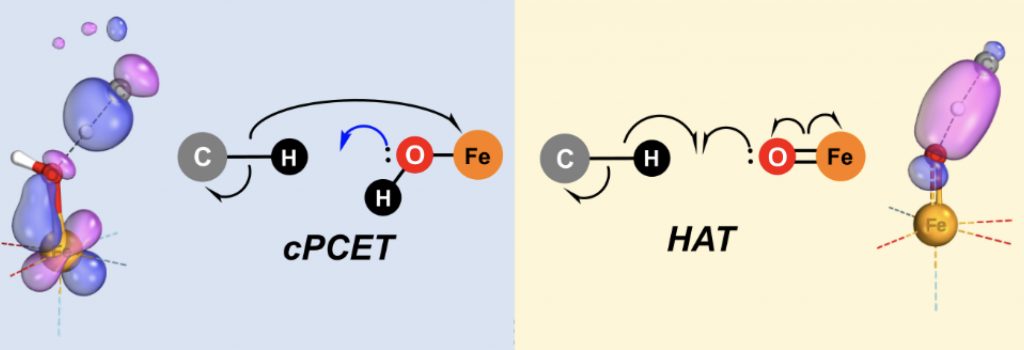
We can see from this that mechanistic reaction analysis is starting to track the “curly arrows” we conventionally use to represent reactions in some detail, as well as informing us about the relative detailing timing of the various curly arrows used. Of course this latter aspect cannot be easily represented by conventional curly arrows. It seems timely to revisit the vast corpus of organic and organometallic “curly arrow pushing” to starting adding such information!
Author
References
- J.E.M.N. Klein, and G. Knizia, "cPCET versus HAT: A Direct Theoretical Method for Distinguishing X–H Bond‐Activation Mechanisms", Angewandte Chemie International Edition, vol. 57, pp. 11913-11917, 2018. https://doi.org/10.1002/anie.201805511
- G. Knizia, "Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts", Journal of Chemical Theory and Computation, vol. 9, pp. 4834-4843, 2013. https://doi.org/10.1021/ct400687b
Tags: chemical reactions, Chemistry, Deprotonation, Hydride, Hydrogen, Hydrogen atom abstraction, Proton, proton travel, Proton-coupled electron transfer, Technology/Internet