In several posts a year or so ago I considered various suggestions for the most polar neutral molecules, as measured by the dipole moment. A record had been claimed[1] for a synthesized molecule of ~14.1±0.7D. I pushed this to a calculated 21.7D for an admittedly hypothetical and unsynthesized molecule. Here I propose a new family of compounds which have the potential to extend the dipole moment for a formally neutral molecule up still further.

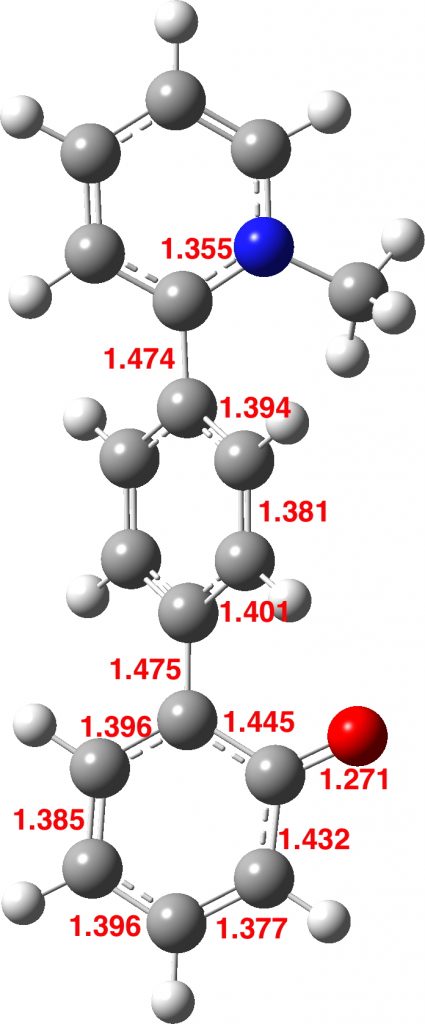
These molecules derive from a well-known class of molecule known as ortho-quinomethides. If the methide part is substituted with an electron donating substituent such as an amino group in 3, a push-pull opportunity now arises, which is strongly driven by aromatisation of the quinomethide ring. This allows one to design “neutral” molecules such as 1 and 2, which now contain respectively two and three rings that will be aromatised by the process. The aromatisation stabilization energy is balanced of course by an opposing increase in energy resulting from charge separation. You can observe that partially aromatising three independent rings as in 2 can drive a great deal of charge separation. One may indeed wonder how much charge separation can be sustained before a triplet instability occurs, driving the molecule back to being neutral. In the case of 2, the wavefunction is in fact stable to such an open shell state, but higher homologues may not be. An aspect worth investing!
| 1 | 2 |
|---|---|
 |
|
| DM 12.3 | DM 31.5 |
| DOI: 10.14469/hpc/4004 | DOI: 10.14469/hpc/4059 |
Molecule 1 does have some precedent in 3[2] but this system exists as a phenol, having abstracted a proton from an acid and leaving behind the acid anion, as per below for 1. Any attempts to deprotonate this phenol with a superstrong base were unreported.

Unsurprisingly therefore, molecules such as 1 and 2 could be regarded as even more highly potent bases than 3, driven again by further aromatisation. The properties of such a potential superbase will be investigated in the next post.
Author
References
- J. Wudarczyk, G. Papamokos, V. Margaritis, D. Schollmeyer, F. Hinkel, M. Baumgarten, G. Floudas, and K. Müllen, "Hexasubstituted Benzenes with Ultrastrong Dipole Moments", Angewandte Chemie International Edition, vol. 55, pp. 3220-3223, 2016. https://doi.org/10.1002/anie.201508249
- N.R. Candeias, L.F. Veiros, C.A.M. Afonso, and P.M.P. Gois, "Water: A Suitable Medium for the Petasis Borono‐Mannich Reaction", European Journal of Organic Chemistry, vol. 2009, pp. 1859-1863, 2009. https://doi.org/10.1002/ejoc.200900056
Tags: aromatisation stabilization energy, Chemical polarity, chemical properties, Chemistry, Dipole, Electric dipole moment, Electromagnetism, energy, Moment, Nature, Physical quantities, Physics, Potential theory
I was wondering why you have preferred to study the ortho compounds rather than the para? Para would seem to give you significantly more charge separation, and the greater torsion of the ortho compounds would seem to reduce the aromatic stabilization more than the linear conjugation effect?
The ortho compound was selected because compound 3 is a known compound and hence there is some precedent. I dare say the para would indeed have even greater charge separation. Perhaps some synthetic chemist out there might be induced to make either or both to see if they can be deprotonated to the highly polar forms?
I spent a quiet lunchtime plotting out the Mulliken charges from your Gaussian.log files attached for 1 & 2 above. I used the figures with hydrogen charges added to the carbon to which they are bonded.
For 2, things come out quite straightforwardly.
The oxygen has a charge of -0.65, offset by +0.22 on it’s attached carbon, and another -0.52 is distributed over the o- and p- carbons, polarized towards the N ring. The m- positions are roughly neutral. Total -0.95.
The central ring is neutral, though slightly polarized towards the O ring.
The nitrogen offloads all its charge, around +0.26 to each adjoining carbon (in fact mainly the hydrogens attached), and +0.12 to the p- carbon. With a tiny contribution from the m- carbons this balances the O ring charge.
For 1 the distribution is more complicated.
The oxygen has -0.94, and its adjacent carbon +0.36, but the rest of the ring is strongly perturbed. The bridgehead carbon has +0.69, a huge charge on that atom, and the other o- carbon a reasonable -0.44. The p- carbon has -0.23. The m- carbon adjacent to the bridgehead has -0.15, presumably attracted there by the +0.69 adjacent. Total -0.67 but a large internal charge separation.
By contrast, the N ring has very low charges. The nitrogen retains +0.28, and its o- carbon away from the bridgehead has +0.45. The other carbons adjacent to nitrogen are nearly neutral. The carbon opposite the bridgehead has -0.12 and the others very little.
So the nitrogen is always good at sharing out its formal positive charge, but I was surprised to find much of it sitting on the O ring bridgehead in 1. Several effects seem to be adding together to produce a complex result here.
And whatever you think of this analysis, many thanks to you for the FAIR data which made it possible for me to carry it out. I am only an amateur chemist, at least since 1974, but I do still find the subject absolutely fascinating.
Thanks for the analysis Mike.
Another way of representing charges is via the MEP (molecular electrostatic potential) field, which simply shows how attractive or repulsive any region of the molecule is to a proton. This contrasts with the atom-centered reduction that atomic charges represent. I should point out that whereas your analysis was able to use the numerical atomic charges in the output, calculating an MEP requires rather more software tools (Gaussview in this case, using the .fchk file instead of the .log file) which may not always be available to interested readers.
The orientation of the dipole comes out quite visually, along with the observation that whilst the π-cloud for the oxygen bearing ring is indeed the -ve part, the top two rings bear the positive charge.
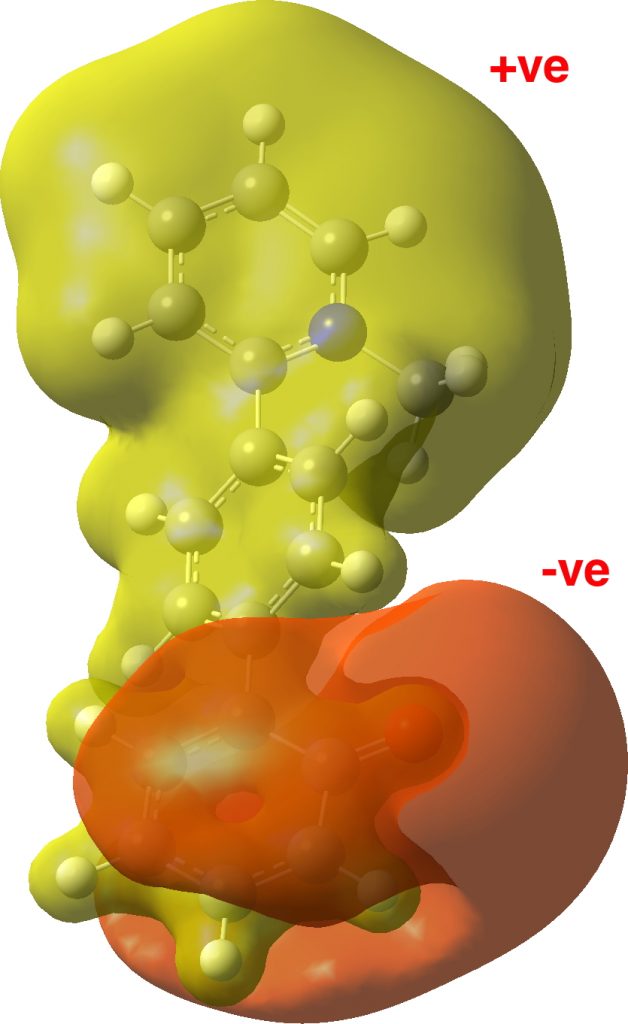
The comments about your being able to use the data for your own analysis are also very timely. ChemistryWorld has a piece coming out any time now (rsc.li/2HG7lTk) on precisely this aspect.
Here is yet another take on the charges, the LUMO (lowest unoccupied molecular orbital) and the HOMO (highest occupied). Both are π-systems, and match the MEP above.
Thankyou for these pictures, they really bring the chemistry to life. I wonder if you could be persuaded to post those for 1 as well? The interactions between the rings look rather interesting.
In case anyone’s interested, I came across this paper on the synthesis of p-quinone methides. Their method wouldn’t directly produce the para analogue of 1, but some fiddling could probably get there. It does prove they are not inaccessible.
http://evans.rc.fas.harvard.edu/pdf/evans38.pdf
Here are the properties for 1, as requested.
Many thanks for these pictures. What has been interesting me is the bridgehead carbon on the O ring. In my original comment, I highlighted that the carbons ortho to the carbonyl have charges of +0.69 and -0.34. The carbonyl has +0.36. Looking at the MEP above, the negative zone clearly has a bite taken out of it, where the bridgehead carbon is. And looking at the structure, the bridgehead – carbonyl bond is the longest one in either ring, which supports the thought that its the best part of an electron short. So I believe there is a real phenomenon here, not an artifact of the localization function generating the Mulliken charges.
But looking at the HOMO, which is overwhelmingly on the O ring, there is an almost perfect symmetry about the C=O axis. And the LUMO, which has some presence on the O ring, is not compellingly asymmetrical.
So what is the basis for this big positive charge on the bridgehead carbon?
In 2, the corresponding carbon relative to the N ring (i.e. the middle ring carbon nearest the N ring) is neutral. The bridgehead carbon in the O ring has -0.11, the other ortho carbon -0.23. So there’s something going on, below the HOMO-LUMO level, to produce a huge interaction between the rings in 1.
Or am I just picking nits in an irritating way?
Dear Prof. Rzepa,
This molecule is very promising.
Another idea for increasing the dipole moment of a molecule is the control of the orientation of each polar monomer in a polymeric chain. I have done a small theoretical study in the paper given as:
DOI: 10.1021/acs.macromol.8b00215
but obviously it is focused on a small number of polymers.
The real problem is the synthesis of such a polymer. Is it possible to obtain polymers where each monomer is oriented in a predefined orientation with respect to its neighboring monomers to achieve a predefined value of dipole moment?
Best regards,
George
P.S. Can you add the vector of the dipole moment in an additional picture?
Here is an image of the dipole vector.
Thanks a lot.
So it is not completely parallel to the backbone axis.
Best,
George